Technology tamfitronics
Main
The transcription factor nuclear factor E2-related factor 2 (NRF2) is an attractive therapeutic target because its activation stimulates a panoply of antioxidants that naturally resolve oxidative stress, a process central to diseases with great societal burden1,2,3,4. However, systemically distributed activators such as monomethyl fumarate (MMF) have global on-target actions because of the ubiquitous expression of NRF2 (refs. 2,3,5,6). In addition to activating NRF2 by modifying Kelch-like ECH-associated protein 1 (KEAP1) thiols (Cys151), MMF also exerts undesirable off-target actions by indiscriminately modifying other protein thiols as well3,4,5,6,7,8. We sought to overcome these dose-limiting and harmful side effects3,4,5,6,9,10 by creating a prodrug that would release MMF only when and where required.
Hydrogen peroxide and peroxynitrite are overproduced at sites of oxidative stress and are ideal molecular triggers for localized prodrug activation as they are restricted to sites of pathology by their inherent reactivity4,11,12. With this in mind, we functionalized MMF with a biologically compatible 1,2-dicarbonyl group (compounds 1a–c; Fig. 1a), designed to preferentially release MMF (with acid 3 as the only byproduct) on specific Baeyer–Villiger oxidation by these peroxides13,14. Peroxides were previously used to trigger prodrug activation in vitro but this reaction was not exploited to safely treat disease through targeted drug release at sites of pathology15,16.

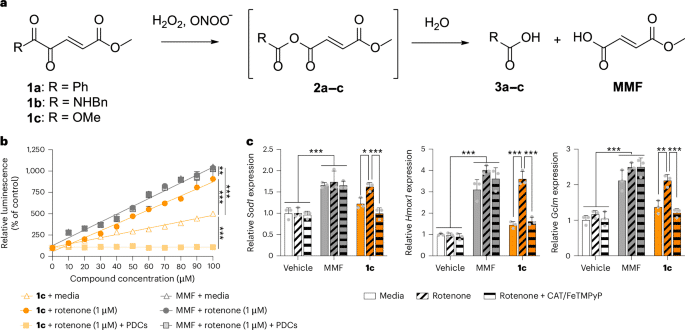
a1,2-Dicarbonyl prodrugs of MMF and their reaction with hydrogen peroxide and peroxynitrite. bCompound 1c increased NRF2 reporter activity when peroxides were endogenously induced in vitro by mitochondrial complex I inhibitor rotenone and was blocked by PDCs. MMF had nonspecific action (n = 3 per group). Extra sum-of-squares F-tests: **P = 0.011 and ***P < 0.001. cCompound 1c (30 μM) increased NRF2 target gene expression in the presence of rotenone and was blocked by PDCs. MMF had nonspecific action (n = 3 per group). Two-way ANOVA with Tukey’s post hoc tests: Sod1*P = 0.036 and ***P < 0.001; Hmox1***P < 0.001; Gclm**P = 0.003 and ***P < 0.001. All data are the mean ± s.d.; individual replicates are presented as gray dots.
Full size image
We synthesized putative 1,2-dicarbonyl prodrugs of MMF (compounds 1a–c) covering three distinct chemotypes (1,2-diketones, α-ketoamides and α-ketoesters; Fig. 1a and Supplementary Fig. 1). These compounds were screened in vitro for their ability to preferentially activate NRF2 upon exposure to peroxides (Supplementary Fig. 2). The α-ketoester 1c enhanced NRF2 reporter activity and expression of target genes encoding antioxidants when peroxides were endogenously overproduced by inhibiting mitochondrial complex I (Fig. 1b,c; P < 0.001). Compound 1c similarly activated NRF2 in the presence of pathological concentrations of exogenous hydrogen peroxide or peroxynitrite (Extended Data Fig. 1a,b; P < 0.001). Physiological concentrations of exogenous peroxides were insufficient for 1c to activate NRF2 (Extended Data Fig. 1c,d; P = 0.087). Decomposing endogenous peroxides with catalase and iron(III) porphyrin complex, FeTMPyP, prevented 1c from activating NRF2 and increasing expression of genes encoding antioxidants (Fig. 1b,c; P < 0.001). These peroxide decomposition catalysts (PDCs) also abolished NRF2 activity induced by 1c per se (Extended Data Fig. 1e; P < 0.001), suggesting that in vitro accumulation of metabolically derived peroxides activates 1c. These results show that peroxides preferentially activate 1c under pathological conditions. To validate MMF as the active metabolite of 1cwe evaluated α-ketoester 5bwhich releases monomethyl succinate upon exposure to peroxides. α-Ketoester 5b failed to activate NRF2 in vitro (Supplementary Fig. 3; P = 0.665), consistent with monomethyl succinate lacking the reactive α,β-unsaturated system through which MMF modifies KEAP1 thiols (thus activating NRF2). Nonfunctionalized MMF activated NRF2 independently of peroxides, whether endogenous (Fig. 1b,c; P = 0.958) or exogenous (Extended Data Fig. 1a,b; P = 0.256). Thus, the 1,2-dicarbonyl functional group of 1c dictates that it preferentially activates NRF2 in the presence of pathological peroxides.
HPLC analysis of the 1c mechanism of action demonstrated that MMF was released cleanly upon exposure to 1.1 and 5 molar equivalents of hydrogen peroxide (Extended Data Fig. 2a). Consistent with the stoichiometry of Baeyer–Villiger oxidation, 0.01 molar equivalents of hydrogen peroxide did not trigger MMF release (Extended Data Fig. 2a). As summarized in Extended Data Fig. 2b, 1H and 13C nuclear magnetic resonance (NMR) analyses showed that 1c established a dynamic equilibrium between the keto and hydrate forms in aqueous solutions17 (Extended Data Fig. 2c and Supplementary Fig. 4). α-Ketoester 1c hydrolyzed to α-ketoacid 1d in phosphate buffer (pH 7.4), which resulted in an equilibrium between the corresponding keto and hydrate forms (Extended Data Fig. 2c). Incubating 1c with two molar equivalents of H2O2 in phosphate buffer (pH 7.4) gave MMF, with complete consumption of 1c after 86.1 min (Extended Data Fig. 2d). These results demonstrate that 1c is susceptible to Baeyer–Villiger oxidation to release MMF upon exposure to biological peroxides.
The fate of 1c after oral administration to rats was next assessed (scheme presented in Extended Data Fig. 3a). α-Ketoester 1c was undetectable in circulation, with only negligible levels of the predicted metabolite α-ketoacid 1d (Extended Data Fig. 3b and Supplementary Table 1). Instead, α-ketoacid glutathione S-conjugate 4d was the predominant metabolite over low levels of α-ketoester glutathione S-conjugate 4c (Extended Data Fig. 3 and Supplementary Table 1). Plasma levels of MMF were subtherapeutic7,8consistent with homeostatic peroxide levels in this compartment (Extended Data Fig. 3b and Supplementary Table 1). Using ultraviolet–visible light spectroscopy, we determined that 1d readily conjugates with glutathione (Supplementary Table 2). Glutathione S-conjugate 4d did not react appreciably with other biological thiols, as shown by mass spectrometry (MS) (Supplementary Table 3). Reaction of two molar equivalents of aqueous hydrogen peroxide with 4d produced glutathione S-conjugate 12 (Supplementary Fig. 1d), a known intermediate in the metabolism of orally administered dimethyl fumarate to MMF18. Together, these data indicate that orally administered 1c undergoes presystemic hydrolysis to give 1d. α-Ketoacid 1d and glutathione rapidly form an equilibrium that favors the formation of nonthioreactive 4d. Glutathione S-conjugate 4d is the major circulating species and is susceptible to Baeyer–Villiger oxidation by biological peroxides, ultimately releasing MMF.
The ability of 1c to preferentially release MMF at localized sites of oxidative stress was investigated in vivo using a mouse model of unilateral peripheral nerve injury (PNI). This injury induces oxidative stress at the sciatic nerve injury site and ipsilateral L4 and L5 dorsal root ganglia (DRG) (Fig. 2a)11,12. Compound 1c induced NRF2 nuclear translocation (activation), measured as a proxy for MMF release, only at the sciatic nerve injury site (Supplementary Fig. 5; P < 0.001) and ipsilateral DRG (Fig. 2b and Supplementary Fig. 6; P < 0.001). Localized NRF2 translocation was accompanied by the increased expression of NRF2 target genes in the ipsilateral DRG (Extended Data Fig. 4a; P < 0.05). These effects were blocked by the coadministration of PDCs (Fig. 2c, Extended Data Fig. 4b and Supplementary Fig. 7; P < 0.01), confirming that biological peroxides are required for activation of NRF2 by 1c. MMF was also verified as the therapeutic metabolite of 1cas substitution for 5b (releasing monomethyl succinate) did not activate NRF2 (Supplementary Fig. 8; P < 0.001). Intravenous administration of glutathione S-conjugate 4dthe circulating metabolite of 1cwas sufficient to preferentially activate NRF2 in the ipsilateral DRG (Extended Data Fig. 4c), consistent with the susceptibility of this species to Baeyer–Villiger oxidation. In contrast to these localized actions of oral 1c and intravenous 4dequimolar doses of oral diroximel fumarate (DRF; prodrug that releases MMF systemically5) indiscriminately activated NRF2 and increased target gene expression in both ipsilateral and contralateral tissues (Fig. 2b,c, Extended Data Fig. 4a and Supplementary Figs. 5, 6 and 7), as well as other organs (Extended Data Fig. 4d; P < 0.001). Intravenous MMF administration had similarly indiscriminate activity in contralateral DRG (Extended Data Fig. 4c). These systemic actions of MMF may interfere with physiological redox signaling and lead to side effects3,4,5,6,9,10.

aSchematic of PNI and tissues of interest. bCompound 1c preferentially increased NRF2 nuclear translocation in ipsilateral L4 and L5 DRG, in contrast to DRF (DRG from three mice pooled per sample). cPDCs abolished NRF2 nuclear translocation induced by 1c in ipsilateral L4 and L5 DRG (DRG from two mice pooled per sample). dIn contrast to DRF, 1c did not reduce plasma glutathione levels (n = 3 mice per sex per group). One-way ANOVA with Tukey’s post hoc test: ***P < 0.001. eIn contrast to DRF, 1c did not increase skin temperature (n = 4 mice per sex per group). One-way ANOVA with Tukey’s post hoc test: **P = 0.002 and ***P < 0.001. fOral 1c and DRF reversed punctate allodynia 6 months after nerve injury (n = 4 mice per sex per group). Repeated-measures two-way ANOVA with Tukey’s post hoc tests: relative to vehicle, *P = 0.029 and ***P < 0.001; 1c versus DRF, #P = 0.012, ##P = 0.003 and ###P < 0.001. BL, baseline. gOral 1c and DRF reversed ongoing pain in the conditioned place preference assay (n = 3 mice per sex per group). Two-way ANOVA with Tukey’s post hoc test: *P = 0.018, **P = 0.007 and ***P < 0.001. hOral treatment with 1c did not reverse punctate allodynia in nerve-injured Nfe2l2−/− mice compared to wild-type controls (n = 4 mice per sex per group). Repeated-measures two-way ANOVA with Tukey’s post hoc test: ***P < 0.001. iOral 1c attenuated punctate allodynia in a model of osteoarthritis (n = 4 mice per sex per group). Repeated-measures two-way ANOVA with Šídák’s post hoc test: *P < 0.05 and **P = 0.002. Compound 1c and DRF were orally dosed at 350 μmol kg−1 day−1represented by gray boxes. Data are the mean ± s.d. Individual replicates are presented as spaghetti plots or gray dots.
Source data
Full size image
Clinical side effects of the systemic distribution of MMF include glutathione depletion and cutaneous flushing5. Consistent with this, in mice treated with DRF, serum glutathione levels were reduced (Fig. 2d; P < 0.001) and skin temperature was increased (a proxy measure of vasodilation that causes flushing5) (Fig. 2e and Supplementary Fig. 9; P < 0.001). Localized release of MMF from 1c should prevent or reduce these side effects. Indeed, glutathione levels (Fig. 2d; P = 0.474) and skin temperature (Fig. 2e and Supplementary Fig. 9; P = 0.482) were unaltered by oral treatment with equimolar doses of 1c compared to vehicle.
PNI often causes chronic neuropathic pain, which is refractory to treatment19,20. Systemically distributed MMF alleviates neuropathic pain in preclinical models by reducing oxidative species that ultimately hyperexcite neurons in pain pathways5,11,12,21,22. We evaluated 1c in the PNI model to determine whether local release of MMF is sufficient to alleviate clinically relevant pain behaviors19. Multiday oral administration of 1c beginning 6 months after nerve injury (a time point capturing clinically relevant pathophysiological mechanisms of neuropathic pain19) reversed hypersensitivity to innocuous punctate, dynamic and cold stimuli (allodynia) (Fig. 2f and Extended Data Fig. 5a,b; P < 0.001). These effects were dose dependent (Extended Data Fig. 5c,d; P < 0.001). Pain relief was also maintained upon repeated dosing (Extended Data Fig. 5e; P < 0.001), indicating that 1c does not induce tolerance, a major limitation of powerful painkillers such as morphine12. Compound 1c further reversed spontaneous and paroxysmal pain induced by nerve injury, assessed with the conditioned place preference paradigm (Fig. 2g; P = 0.001). Importantly, 1c retained or exceeded the pain-relieving effects of DRF at equimolar doses (Fig. 2f,g and Extended Data Fig. 5a–d). Normal sensory function was spared by 1cas responses to punctate (Extended Data Fig. 5c; P = 0.460) and dynamic stimuli (Extended Data Fig. 5d; P = 0.242) were unaltered in sham-operated animals. Compound 1c failed to reverse pain in nerve-injured NRF2-deficient mice (Fig. 2h; P < 0.001), which indicates that NRF2 is the therapeutic target of 1cconsistent with the antinociceptive mechanism of MMF21. The knockout did not confound the assessment of analgesia, as morphine still relieved allodynia (Extended Data Fig. 5f,g; P < 0.001). Moreover, MMF was confirmed as the therapeutic metabolite of 1cas 5b did not reverse nerve injury-induced allodynia (Supplementary Fig. 10; P = 0.864). Intravenous administration of the circulating metabolite glutathione S-conjugate 4d was also sufficient to reverse allodynia (Extended Data Fig. 5h), replicating the pharmacodynamics of oral 1c administration. The pain-relieving effects of 1c broadly extended to other models of chronic pain that feature oxidative stress11,12 such as osteoarthritis (Fig. 2i; P < 0.001), chemotherapy-induced peripheral neuropathy (CIPN; Extended Data Fig. 5i,j; P < 0.001) and diabetic neuropathy (Extended Data Fig. 5k; P = 0.002).
We present a 1,2-dicarbonyl prodrug that leverages Baeyer–Villiger oxidation by pathological peroxides to preferentially release MMF at localized sites of oxidative stress. This approach suppresses the systemic action of MMF. Our prodrug has therapeutic efficacy in pathological conditions, relieving chronic pain in established preclinical models. Chronic pain remains a large unmet medical need and nonaddictive treatments engaging endogenous pain resolution mechanisms such as NRF2 are a unique approach20,22. The localized release of MMF reduces unwanted systemic NRF2 activation and related side effects, which have until now limited the therapeutic potential of this transcription factor2,3,4,5. Whether localized MMF release similarly reduces the risk of NRF2-dependent chemoresistance and progression of some cancers should be carefully evaluated2,23. The 1,2-dicarbonyl platform presented here is a candidate drug class that is biologically compatible and stable, has broad synthetic scope and is widely applicable to numerous other diseases and disorders of oxidative stress2,3,4,24.
Methods
Synthesis and characterization of 1,2-dicarbonyl compounds 1a–d, glutathione S-conjugates 4c, 4d and 12 and MMF
Details of the synthesis and full compound characterization are presented in the Supplementary Methods. In brief, 1,2-dicarbonyl 1a was prepared from cinnamaldehyde 6 (Supplementary Fig. 1b) in three steps. α-Keto amide 1b was synthesized as reported previously25 (Supplementary Fig. 1a). α-Ketoester 1c was synthesized from dimethyl 2-oxoglutarate 5busing the similar bromination and elimination conditions described for 1b (Supplementary Fig. 1a)25. Phosphate buffer-mediated hydrolysis of 1c gave α-ketoacid 1d (Supplementary Fig. 1a). Glutathione S-conjugates 4c and 4d were synthesized from 1c and 1drespectively, upon reaction with glutathione in a mixture of water and acetonitrile (Supplementary Fig. 1c). Reaction of 4d with aqueous hydrogen peroxide gave compound 12 (Supplementary Fig. 1d), a conjugate of glutathione with MMF. MMF was synthesized from maleic anhydride as reported previously26 (Supplementary Fig. 1e). Compound 1a and MMF were stored at ambient temperature with protection from light and compounds 1b–d, 4c, 4d and 12 were stored as solids below 4 °C in the absence of light.
Reaction of 1c with hydrogen peroxide
Analytical reverse-phase high performance liquid chromatography (HPLC) spectra of MMF, 1c and the reactions of 1c with hydrogen peroxide were acquired on an Agilent 1100 Infinity series machine using a Phenomenex Luna Omega PS C18 column (250 × 4.6 mm, 100 Å, 5 μm) with a buffer system of 0.1% trifluoroacetic acid (TFA) in water (buffer A) and 0.1% TFA in acetonitrile (buffer B). Elution was conducted with a gradient of 0–100% A–B over 15 min, using a flow rate of 1.0 ml min−1. To a solution of 2,778 µM, 611 µM or 5.56 µM hydrogen peroxide in water (900 µl) was added a 5 mM solution of 1c in water (100 µl) to give a final concentration of 500 µM 1c and 2,500 µM (5 equivalents), 550 µM (1.1 equivalents) or 5 µM (0.01 equivalents) hydrogen peroxide. After 40 and 130 min, the samples were analyzed using HPLC.
Compound 1c was reacted with hydrogen peroxide and the rate of MMF formation was measured by NMR spectroscopy of a sample of 1c (4.30 mg, 0.025 mmol, 1.0 equivalent) and 2,2-dimethyl-2-silapentane-5-sulfonate-d6 sodium salt (DSS-d6) (1.12 mg, 0.005 mmol) in a mixture of 111 mM phosphate buffer (pH 7.4; 450 µl) and D2O (50 µl). Then, 30% w/w hydrogen peroxide solution (5.11 µl, 0.050 mmol, 2.0 equivalents) was added to initiate the reaction. 1H NMR spectra of 1c in 100 mM phosphate buffer (pH 7.4) or water and MMF (synthesized by the authors) in 100 mM phosphate buffer (pH 7.4) were taken at a concentration of 50 mM 1c or MMF and referenced with DSS.
Reaction of 1d with glutathione
First, 100 µl each of stock solutions of 1d (20 mM) and glutathione (40 mM) in 100 mM phosphate buffer (pH 6.0, 7.0, 7.4 and 8.0) were combined in a 96-well plate to give 200 μl of reaction mixture containing 10 mM 1d and 20 mM glutathione. The absorbance at 279 nm was monitored over time using a BioTek Synergy H4 Hybrid plate reader and the resulting data were normalized to the absorbance of a 10 mM solution of 1d at the respective pH. Pseudo first-order rate constants, k (min−1), were calculated as the exponents of exponential least-squares regression functions.
Reaction of 4d with N-acetylcysteine
A 1 mM solution of 4d (0.2 ml, 0.0002 mmol) and a 1 mM solution of N-acetylcysteine (0.2 ml, 0.0002 mmol) were added to water (0.6 ml). The count of the [M + H]+ ions for each species was measured at 0.5 h and 16 h by electrospray ionization MS in positive mode on a Bruker HCT MS instrument.
In vitro NRF2 reporter assay
NRF2/ARE luciferase reporter HEK293 cells (SL-0042-NP, Signosis) were maintained in DMEM (SH30243.01, Cytiva) supplemented with 10% FBS (03-600-511, Thermo Fisher Scientific) and 1% penicillin–streptomycin (SV30010, Cytiva). Cells were seeded into 48-well flat-bottom cell culture plate (3548, Corning) in 500 μl of supplemented DMEM at a concentration of 2 × 105 cells per ml and incubated overnight at 37 °C with 5% CO2 in a humidified environment. Before treatments, the medium was replaced with fresh 0.1% FBS in DMEM. tert-Butylhydroquinone (0–30 μM; 112941, Millipore Sigma), a well-known NRF2 activator, was used as a positive control to confirm NRF2-dependent luciferase production (Supplementary Fig. 11).
Cells were treated with a concentration range of rotenone (mitochondrial complex I inhibitor; R8875, Millipore Sigma) (0–100 μM) to identify the concentrations that did not activate NRF2 per se and were not cytotoxic (Supplementary Fig. 12). In separate experiments, cells were treated with concentration ranges of 1c or MMF (651419, Millipore Sigma) (1–100 μM) followed by rotenone (1 μM) to endogenously overproduce peroxides27. The medium was used as the negative control for all conditions.
To identify concentrations of hydrogen peroxide and peroxynitrite that did not activate NRF2 per se, cells were treated with concentration ranges of hydrogen peroxide (H1009, Millipore Sigma) or peroxynitrite (20-107, Millipore Sigma) (0–300 μM) (Supplementary Fig. 11). In separate experiments, cells were treated with concentration ranges of 1a–c, 5b (349631, Millipore Sigma) or MMF (651419, Millipore Sigma) (1–100 μM)7,8,28followed by a fixed concentration of exogenous hydrogen peroxide (1 or 10 μM) or peroxynitrite (1 or 20 μM). These peroxide concentrations represent those in the physiological and pathological range, respectively29,30. The medium alone was used as the negative control for all conditions.
For experiments to eliminate peroxides, cells were treated with a concentration range of 1c (0–100 μM), together with 250 U per ml catalase (E3289, Millipore Sigma)31 and 50 μM FeTMPyP (75854, Cayman Chemical)32followed by rotenone (1 μM).
Cells were incubated with treatments for 4 h for studies of gene expression or 16 h for luciferase reporter activity. To quantify luciferase activity, cells were washed with PBS (Thermo Fisher Scientific) and then lysed by a 15-min incubation at room temperature with passive lysis buffer (E1941, Promega). Cell lysates (30 μl) were transferred to a 96-well white or clear flat-bottom plate (3632, Corning) and mixed with 150 μl of luciferase substrate (LUC100, Signosis). The plates were read in a Synergy HTX multimode reader (BioTek). All conditions were performed in triplicate and observations were verified across three independent experiments.
Animals
Pathogen-free adult male and female C57Bl/6J (8 weeks old on arrival; strain 000664) and C57Bl/6J diet-induced obese (DIO) (8 weeks old on arrival; strain 380050) mice were purchased from The Jackson Laboratory (Bar Harbor). Male and female (8–12 weeks old) Nfe2l2−/− mice and wild-type littermates on a C57BL/6J genetic background (The Jackson Laboratory; strain 017009) were bred at The University of Texas MD Anderson Cancer Center. Male Sprague–Dawley rats were purchased from Inotiv. Mice were housed 5 per cage and rats were housed 3–4 per cage in a light-controlled and temperature-controlled room (12-h light–dark cycle; lights on at 7:00 a.m.) with food and water available ad libitum. Equal numbers of male and female rodents were used in each experimental group, except for pharmacokinetic studies (males only). Animals were randomly assigned to experimental groups using a randomization calculator (GraphPad Software). All animals were acclimated to the animal care facility for at least 7 days before the start of the study. Procedures were approved by the Animal Care and Use Committees of MD Anderson Cancer Center and Texas Southern University.
Surgical PNI
Spared nerve injury33 was performed in mice34. Briefly, under inhaled isoflurane anesthesia, the tibial and common peroneal nerves were isolated, tightly ligated with 6–0 silk (707G, Ethicon), and transected immediately distal to the ligation. The sural nerve was left intact. For sham surgery, the nerves were exposed but not ligated or transected. Animals were monitored postoperatively until fully ambulatory before return to their home cage.
Surgical destabilization of the medial meniscus (DMM)
DMM was performed to induce osteoarthritis35. Mice were anesthetized with isoflurane and placed in dorsal recumbency. Carprofen (5 mg kg−1subcutaneously (s.c.)) was administered immediately before surgery began. A 3-mm longitudinal incision was made over the right patella and the joint capsule was opened with micro Vannas scissors under ×4 magnification. Blunt dissection of the infrapatellar fat pad exposed the meniscotibial ligament of the medial meniscus, which was then transected using a no. 11 scalpel blade. The incision was closed with two 9-mm AutoClip wound clips, which were removed 10 days after surgery. Animals were monitored postoperatively until fully ambulatory before return to their home cage.
CIPN
Cisplatin (TEVA Pharmaceuticals) was diluted in sterile saline and administered for 5 days (2.3 mg kg−1 day−1intraperitoneally (i.p.)) followed by 5 days of rest and a second round of five doses to induce CIPN36.
High-fat diet (HFD)-induced neuropathy
Upon arrival, DIO mice were fed an HFD (Research Diets D12492 (60% kcal from fat) to induce type 2 diabetic neuropathy37. Mice were maintained on an HFD for the duration of the study, with drug dosing beginning 2 weeks after arrival.
In vivo drug treatments
Compound 1c (molecular weight (MW) = 172.14) and DRF (MW = 255.23) were suspended in methylcellulose (viscosity: 15 cP, 2% w/v in water; Millipore Sigma). Compound 4d (MW = 465.44) and MMF (MW = 130.10) were dissolved in 0.9% saline. For pharmacokinetic assessments, 1c (350 μmol kg−1orally (p.o.)) was administered as a single dose. For assays of glutathione levels, mice were treated daily with 1c (350 μmol kg−1 day−1p.o.), DRF (350 μmol kg−1 day−1p.o.) or methylcellulose vehicle for 3 days, beginning 7 days after PNI. For thermal imaging, 7 days after PNI, mice were treated with a single dose of 1c (350 μmol kg−1 day−1p.o.), DRF (350 μmol kg−1 day−1p.o.) or methylcellulose vehicle.
For PNI antinociception, 1c (350 μmol kg−1 day−1p.o.), DRF (350 μmol kg−1 day−1p.o.) or vehicle control (equal volume, p.o.) was administered once daily for 7 days, beginning 180 days after PNI. For other PNI antinociception and postmortem tissue analyses, 1c (100, 225 or 350 μmol kg−1 day−1after), 5b (350 μmol kg−1 day−1p.o.), DRF (350 μmol kg−1 day−1after), 4d (190 μmol kg−1 day−1intravenously (i.v.)), MMF (190 μmol kg−1 day−1i.v.) or an equal volume of vehicle control (p.o. or i.v., as appropriate) was administered once daily, beginning 7 days after PNI or sham surgery and continuing for 3 days (postmortem studies and reflex tests) or 7 days (conditioned place preference tests). For other postmortem tissue analyses after peroxide decomposition, catalase (250 U, i.t.)38 and FeTMPyP (5 mg kg−1i.p.)39 or the vehicle control was administered 30 min before and 60 min after once-daily 1c (350 μmol kg−1 day−1p.o.), DRF (350 μmol kg−1 day−1p.o.) or vehicle control (equal volume, p.o.), beginning 7 days after PNI or sham surgery and continuing until day 9.
For DMM antinociception, 1c (350 μmol kg−1 day−1p.o.) or methylcellulose vehicle was administered for five consecutive days, beginning 4 weeks after surgery. For antinociceptive tolerance, 1c (350 μmol kg−1 day−1p.o.) or the positive control morphine sulfate (3 mg kg−1 b.i.d., s.c.; gifted from the National Institute on Drug Abuse Drug Supply Program, Research Triangle Institute) was administered daily for 5 days, beginning 7 days after PNI. For acute morphine analgesia, morphine sulfate (5 mg kg−1s.c.) or saline vehicle was administered once in a randomized crossover design between 19 and 22 days after PNI. For CIPN antinociception, 1c (350 μmol kg−1 day−1p.o.) or vehicle was administered for five consecutive days, beginning 3 days after the last cisplatin dose. For HFD-induced neuropathy antinociception, 1c (225 μmol kg−1 day−1p.o.) or vehicle was administered for five consecutive days, beginning 4 weeks after initiation of the HFD.
In vivo pharmacokinetics
To facilitate the withdrawal of multiple, timed blood draws for pharmacokinetic analysis, the right jugular vein of each rat was cannulated 1 day before drug administration. Under anesthesia (ketamine, acetopromazine and xylazine, 50:3.3:3.3 mg kg−1i.p.), silicone elastomer tubing (0.02 × 0.037 inches) was inserted into the jugular vein, secured with a silk suture and exteriorized in the dorsal infrascapular region. The surgical incision was prophylactically treated with nitrofurazone wound powder and closed with surgical staples. The cannulae were flushed daily with 0.5 ml of sterile heparinized saline (100 U per ml). All studies were initiated between 8:00 a.m. and 9:00 a.m. to eliminate possible circadian variation. The animals were fasted overnight and until 4 h after dosing, with water available ad libitum. Blood samples (0.15 ml) were collected from the cannulae immediately before administration of 1c and at 0.083, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6 and 8 h after dosing. Plasma samples were isolated, immediately mixed with formic acid (1:1 v/v), stored at −80 °C and analyzed within 1 week using LC–MS/MS. Concentrations were calculated from standard curves of each analyte in rat plasma.
Tissue collection
Within 4 h of the final dose, mice were deeply anesthetized with Beuthanasia-D (Merck) and then transcardially perfused with ice-cold saline. In some experiments, blood was collected by cardiac puncture before perfusion. The ipsilateral and contralateral sciatic nerve (5 mm proximal to the transection), L4 and L5 DRG, heart, liver, kidney and lung were isolated and rapidly frozen for subsequent analysis.
qPCR
Total RNA was extracted from HEK293 cells or DRG tissues using TRIzol (Thermo Fisher Scientific). Then, 1 μg of RNA was used for reverse transcription with iScript reverse transcription supermix (Bio-Rad). Real-time PCR was carried out in a final volume of 20 μl with iTaq Universal SYBR green supermix (Bio-Rad) containing 2 μl of fivefold diluted complementary DNA and monitored by a CFX Connect real-time PCR detection system (Bio-Rad). The following cycling parameters were used: 95 °C for 3 min, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Primer sequences are reported in Supplementary Table 4. The level of the target mRNA was quantified relative to the housekeeping gene (Gapdh) using the DDCt method. Gapdh was not significantly different between treatments.
Western blotting
DRG or sciatic nerves from two or three mice were pooled within groups (treatment, lateralization and sex) to ensure that sufficient protein could be obtained for analysis. Liver, kidney, heart and lung were not pooled. Nuclear fractions were isolated with an NE-PER nuclear and cytoplasmic extraction kit (78835, Thermo Fisher Scientific), according to the manufacturer’s instructions. Western blotting was performed as previously described21. Nuclear proteins were subjected to NuPAGE Bis-Tris (4–12%) gel electrophoresis under reducing conditions. After transfer to nitrocellulose membranes (IB23001, Invitrogen), nonspecific binding sites were blocked with Superblock buffer (37515, Thermo Fisher Scientific) for 1 h at room temperature. Membranes were incubated overnight at 4 °C with primary anti-NRF2 antibody (1:1,000; rabbit polyclonal IgG; ab31163, Abcam) and anti-histone H3 antibody (1:2,000; rabbit polyclonal IgG; ab1791, Abcam) (loading control). The membranes were then washed with PBS containing 0.1% Tween-20 and probed with horseradish peroxidase-conjugated secondary antibody (1:5,000; goat polyclonal IgG; Jackson ImmunoResearch) in blocking buffer containing 0.1% Tween-20 for 1 h at room temperature. After washing with 1× PBS containing 0.1% Tween-20, membranes were developed with enhanced chemiluminescence substrate (Thermo Fisher Scientific). Images were acquired using ImageQuant LAS 4000 (GE Healthcare Life Sciences). Densitometry analysis was performed using ImageQuant TL software (GE Healthcare Life Sciences). Data were normalized to the loading control (histone H3) and then to the vehicle control within each gel.
Glutathione assay
Serum samples were assayed in duplicate for glutathione content (703002, Cayman Chemical), according to the manufacturer’s instructions. The plates were read in a Synergy HTX multimode reader (BioTek).
Thermal imaging
For thermographic analysis of ear skin, mice were housed individually in a small plexiglass enclosure on a mesh stand. Images were acquired between 10:30 and 11:30 a.m. using a thermal camera (C5; Teledyne FLIR), before and 30 min after treatment. The intensity of a single pixel was calculated at the center of the spot meter (Thermal Studio, Teledyne FLIR), obtained within the antihelix of the pinna (region of interest). Three measurements were made in each ear (where possible) and averaged to obtain the skin temperature for each mouse. Data are presented as the difference between the post-treatment and baseline measurements for each mouse. Image acquisition and analysis were performed by an experimenter who was blinded to treatment conditions.
Behavioral sensory testing
All behavioral tests were conducted by an experimenter who was blinded to group assignments. Mice received at least three 60-min habituations to the test environment before reflex testing. Rodents were placed in a small plexiglass enclosure on a mesh stand. Punctate allodynia was measured using the von Frey test21. The 50% paw withdrawal threshold was determined using the ‘up–down’ method40. Dose-dependent reversal of allodynia is reported as a percentage of the maximum possible antiallodynia, calculated for each mouse as a ratio of its actual antiallodynia compared to a hypothetical situation in which the drug brought withdrawal thresholds to their original baseline at the postinjection time point.
Dynamic allodynia was measured by lightly stroking the plantar surface of the hindpaw with a soft paintbrush41. A paintbrush (5/0, Princeton Art and Brush) was prepared by blunting the tip and removing the outer layer of hairs. The lateral plantar region of the left hindpaw (sural nerve territory) was stimulated by light stroking (∼2 cm s−1) with the paintbrush, in the direction from heel to toe. The paw withdrawal response was scored according to the following criteria: score = 0, walking away or occasionally very brief paw lifting (≤1 s); score = 1, sustained lifting (>2 s) of the stimulated paw toward the body; score = 2, a strong lateral lifting above the level of the body; score = 3, flinching or licking of the affected paw. Average scores for each mouse were obtained from three stimulations at intervals of at least 3 min.
Cold allodynia was measured by applying a drop of acetone to the midplantar area of the hindpaw and monitoring the behavioral responses for 1 min42,43. The cumulative time spent in pain behavior (paw licking, withdrawal, flinching, etc.) was recorded. The test was repeated three times at intervals of at least 15 min to obtain an average time in seconds for each animal.
Ongoing pain was tested using a conditioning paradigm44 with retigabine (R-100, Alomone Laboratory) as the conditioned stimulus to briefly relieve pain36,45,46. Mice were first allowed to freely explore the conditioned place preference apparatus, consisting of two chambers (one dark and one light) connected by a hallway (Stoelting), for 15 min. The time spent in the light chamber was recorded. During the conditioning phase, mice were first administered saline (i.p.) and kept in the dark chamber for 20 min. Then, 3 h later, the analgesic retigabine was administered (10 mg kg−1; i.p.) and, after 10 min, the mice were placed in the light chamber for 20 min. The conditioning was completed over four consecutive days. On the fifth day, the mice were again allowed to freely explore the apparatus for 15 min without any retigabine or saline injections. Data are presented as the difference in time spent in the light (retigabine-paired) chamber during the drug-free test on day 5 minus time spent in the light chamber at baseline (preconditioning phase). A mouse with spontaneous pain should show an increase in time spent in the light chamber that was paired with retigabine than it did in the preconditioning phase.
To examine numbness, we used a modified protocol of the adhesive removal test36,47. A round adhesive patch (3/16-inch Teeny Tough-Spots, USA Scientific) was placed on the plantar surface of the hind paws. The latency to attend to the patch (for example, shaking or attempted removal) was recorded within a 15-min testing time.
Statistics
All statistical results are reported in Supplementary Table 5. Differences between in vitro concentration–response relationships were determined by comparing the slopes of fitted linear functions for shared parameters using extra sum-of-squares F-tests. Linear models were selected as the pharmacologically relevant concentrations tested were within the linear range of the concentration–response functions. Differences in gene expression, protein levels and skin temperatures were analyzed by t-tests or one-way or two-way analysis of variance (ANOVA) followed by Dunnett’s or Tukey’s post hoc tests as appropriate. Von Frey and acetone data were analyzed by repeated-measures two-way or three-way ANOVA with Tukey or Šidák’s post hoc tests, one-way ANOVA with Dunnett’s post hoc tests or unpaired t-tests where appropriate. Brush data were analyzed by Friedman with Dunn’s post hoc tests, Kruskal–Wallis with Dunn’s post hoc tests or Mann–Whitney tests where appropriate. Conditioned place preference data were analyzed by two-way ANOVA with Tukey’s post hoc tests. Data from the adhesive removal test were analyzed by unpaired t-test. Analyses were performed using Prism version 10.2.2 (GraphPad). P < 0.05 was considered statistically significant. Parametric data are expressed as the mean ± s.d. and nonparametric data are presented as the median ± range.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data to support the results are available from figshare (https://doi.org/10.6084/m9.figshare.23278331)48. Source data are provided with this paper.
References
GBD Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 3961204–1222 (2020).
Article Google Scholar
Dodson, M. et al. Modulating NRF2 in disease: timing is everything. Annu. Rev. Pharmacol. Toxicol. 59555–575 (2019).
Article CAS PubMed Google Scholar
Cuadrado, A. et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 18295–317 (2019).
Article CAS PubMed Google Scholar
Forman, H. J. &a mp; Zhang, H. Targeting oxidative stress in disease: promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 20689–709 (2021).
Article CAS PubMed PubMed Central Google Scholar
Hoogendoorn, A. et al. Emerging therapeutic applications for fumarates. Trends Pharmacol. Sci. 42239–254 (2021).
Article CAS PubMed PubMed Central Google Scholar
Dinkova-Kostova, A. T. & Copple, I. M. Advances and challenges in therapeutic targeting of NRF2. Trends Pharmacol. Sci. 44137–149 (2023).
Article CAS PubMed Google Scholar
Linker, R. A. et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134678–692 (2011).
Article PubMed Google Scholar
Ahuja, M. et al. Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-induced experimental Parkinson’s-like disease. J. Neurosci. 366332–6351 (2016).
Article CAS PubMed PubMed Central Google Scholar
de Zeeuw, D. et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 3692492–2503 (2013).
Article PubMed PubMed Central Google Scholar
Qin, Q. et al. Nrf2-mediated cardiac maladaptive remodeling and dysfunction in a setting of autophagy insufficiency. Hypertension 67107–117 (2016).
Article CAS PubMed Google Scholar
Grace, P. M. et al. Nitroxidative signaling mechanisms in pathological pain. Trends Neurosci. 39862–879 (2016).
Article CAS PubMed PubMed Central Google Scholar
Squillace, S. & Salvemini, D. Nitroxidative stress in pain and opioid-induced adverse effects: therapeutic opportunities. Pain 163205–213 (2022).
Article CAS PubMed Google Scholar
Abo, M. et al. Development of a highly sensitive fluorescence probe for hydrogen peroxide. J. Am. Chem. Soc. 13310629–10637 (2011).
Article CAS PubMed Google Scholar
Sawaki, Y. & Foote, C. S. Acyclic mechanism in the cleavage of benzils with alkaline hydrogen-peroxide. J. Am. Chem. Soc. 1016292–6296 (1979).
Article CAS Google Scholar
Meng, T. T. et al. Introduction of the alpha-ketoamide structure: en route to develop hydrogen peroxide responsive prodrugs. Chem. Sci. 107156–7162 (2019).
Article CAS PubMed PubMed Central Google Scholar
Lu, M. C., Zhang, X., Zhao, J., You, Q. D. & Jiang, Z. Y. A hydrogen peroxide responsive prodrug of Keap1–Nrf2 inhibitor for improving oral absorption and selective activation in inflammatory conditions. Redox Biol. 34101565 (2020).
Article CAS PubMed PubMed Central Google Scholar
Brun, N., Gonzalez-Sanchez, J. M., Demelas, C., Clement, J. L. & Monod, A. A fast and efficient method for the analysis of alpha-dicarbonyl compounds in aqueous solutions: development and application. Chemosphere 319137977 (2023).
Article CAS PubMed Google Scholar
Dibbert, S., Clement, B., Skak-Nielsen, T., Mrowietz, U. & Rostami-Yazdi, M. Detection of fumarate-glutathione adducts in the portalvein blood of rats: evidence for rapid dimethylfumarate metabolism. Arch. Dermatol. Res. 305447–451 (2013).
Article CAS PubMed Google Scholar
Colloca, L. et al. Neuropathic pain. Night. Rev. Haze. Primary 317002 (2017).
Article PubMed Google Scholar
Price, T. J. et al. Transition to chronic pain: opportunities for novel therapeutics. Nat. Rev. Neurosci. 19383–384 (2018).
Article CAS PubMed PubMed Central Google Scholar
Li, J. et al. Oral dimethyl fumarate reduces peripheral neuropathic pain in rodents via NFE2L2 antioxidant signaling. Anesthesiology 132343–356 (2020).
Article CAS PubMed Google Scholar
Vasavda, C. et al. Identification of the NRF2 transcriptional network as a therapeutic target for trigeminal neuropathic pain. Sci. Adv. 8eabo5633 (2022).
Article CAS PubMed PubMed Central Google Scholar
Lennicke, C., Rahn, J., Lichtenfels, R., Wessjohann, L. A. & Seliger, B. Hydrogen peroxide—production, fate and role in redox signaling of tumor cells. Cell. Commun. Signal 1339 (2015).
Article PubMed PubMed Central Google Scholar
Wang, M., Dai, Z. & Jiang, X. Design and application of alpha-ketothioesters as 1,2-dicarbonyl-forming reagents. Nat. Common. 102661 (2019).
Article PubMed PubMed Central Google Scholar
Laras, Y. et al. Synthesis of quinoline dicarboxylic esters as biocompatible fluorescent tags. J. Org. Chem. 778294–8302 (2012).
Article CAS PubMed Google Scholar
Raillard , SP , Scheuerman , RA & Manthati , SK PCT/US2014/041406 (USA, 2014).
Forkink, M. et al. Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol. 6607–616 (2015).
Article CAS PubMed PubMed Central Google Scholar
Scannevin, R. H. et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 341274–284 (2012).
Article CAS PubMed Google Scholar
Forman, H. J., Bernardo, A. & Davies, K. J. What is the concentration of hydrogen peroxide in blood and plasma? Arch. Biochem. Biophys. 60348–53 (2016).
Article CAS PubMed Google Scholar
Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: redox pathways in molecular medicine. Proc. Natl Acad. Sci. USA 1155839–5848 (2018).
Article CAS PubMed PubMed Central Google Scholar
Preston, T. J., Muller, W. J. & Singh, G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J. Biol. Chem. 2769558–9564 (2001).
Article CAS PubMed Google Scholar
Misko, T. P. et al. Characterization of the cytoprotective action of peroxynitrite decomposition catalysts. J. Biol. Chem. 27315646–15653 (1998).
Article CAS PubMed Google Scholar
Decosterd, I. & Woolf, C. J. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87149–158 (2000).
Article PubMed Google Scholar
Shields, S. D., Eckert, W. A. & Basbaum, A. I. Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J. Pain. 4465–470 (2003).
Article PubMed Google Scholar
Glasson, S. S., Blanchet, T. J. & Morris, E. A. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthr. Cartil. 151061–1069 (2007).
Article CAS Google Scholar
Krukowski, K. et al. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 1581126–1137 (2017).
Article CAS PubMed PubMed Central Google Scholar
Guilford, B. L., Ryals, J. M. & Wright, D. E. Phenotypic changes in diabetic neuropathy induced by a high-fat diet in diabetic C57BL/6 mice. Exp. Diabetes Res. 2011848307 (2011).
Article CAS PubMed PubMed Central Google Scholar
Lu, J. M., Gong, N., Wang, Y. C. & Wang, Y. X. D-Amino acid oxidase-mediated increase in spinal hydrogen peroxide is mainly responsible for formalin-induced tonic pain. Br. J. Pharmacol. 1651941–1955 (2012).
Article CAS PubMed PubMed Central Google Scholar
Doyle, T. et al. Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J. Neurosci. 326149–6160 (2012).
Article CAS PubMed PubMed Central Google Scholar
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 5355–63 (1994).
Article CAS PubMed Google Scholar
Cazuza, R. A. et al. 18 kDa translocator protein (TSPO) is upregulated in rat brain after peripheral nerve injury and downregulated bydiroximel fumarate. Brain Behav. Immun. 12311–27 (2024).
Article PubMed Google Scholar
Carlton, S. M., Lekan, H. A., Kim, S. H. & Chung, J. M. Behavioral manifestations of an experimental model for peripheral neuropathy produced by spinal nerve ligation in the primate. Pain 56155–166 (1994).
Article PubMed Google Scholar
Yoon, C., Wook, Y. Y., Sik, N. H., Ho, K. S. & Mo, C. J. Behavioral signs of ongoing pain and cold allodynia in a rat model of neuropathic pain. Pain 59369–376 (1994).
Article PubMed Google Scholar
King, T. et al. Unmasking the tonic-aversive state in neuropathic pain. Nat. Neurosci. 121364–1366 (2009).
Article CAS PubMed PubMed Central Google Scholar
Blackburn-Munro, G. & Jensen, B. S. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur. J. Pharmacol. 460109–116 (2003).
Article CAS PubMed Google Scholar
Yang, Q. et al. Persistent pain after spinal cord injury is maintained by primary afferent activity. J. Neurosci. 3410765–10769 (2014).
Article PubMed PubMed Central Google Scholar
Bouet, V. et al. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat. Protoc. 41560–1564 (2009).
Article CAS PubMed Google Scholar
Avery, T. D. et al. Peroxide-triggered release of monomethyl fumarate at sites of pathology. figshare https://doi.org/10.6084/m6089.figshare.23278331 (2024).
Download references
Acknowledgements
This work was supported by the National Institutes of Health (grants UG3NS127251 (A.D.A. and P.M.G.), RF1NS113840 (P.M.G.), U54MD007605 (H.X. and D. L.) and in part P30CA016672 (animal housing and care in the MD Anderson Research Animal Support Facility)), the Cancer Prevention Research Institute of Texas (RP180748; H.X. and D. L.), the Australian Research Council (grants CE140100003 and DP180101581; A.D.A.) and a Rita Allen Foundation Award in Pain (A.J.S.). Figure 2a was created with BioRender.com. We thank A. Kavelaars for her insightful comments on a draft of this paper.
Ethics declarations
Competing interests
T.D.A., J.L., D.J.L.T., A.D.A. and P.M.G. are named inventors on patent application WO/2021/179049 and provisional patent application 63/641,731 covering peroxide-activated prodrugs to treat disorders of oxidative stress. T.D.A, A.D.A. and P.M.G. received funding from Biogen. T.D.A., D.J.L.T., A.D.A. and P.M.G. founded and/or hold equity in ImmunoLogic, a start-up company that is developing pathology-activated prodrugs to treat diseases of oxidative stress. The other authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Compound 1c preferentially activated NRF2 in vitro in the presence of pathological levels of exogenous peroxides.
(a) Compound 1c (10-100 μM) enhanced NRF2 activity in the presence of hydrogen peroxide and peroxynitrite, at levels comparable to monomethyl fumarate (MMF). N = 3/group. Extra sum-of-squares F tests, not significant (n.s.), ***P < 0.001. Data are mean ± s.d. (b) In contrast to MMF (30 μM), compound 1c (30 μM) preferentially increased NRF2 target gene expression in the presence of pathological peroxide concentrations. N = 3/group. Two-way ANOVA with Tukey’s post hoc tests, **P < 0.01, ***P < 0.001. Data are mean ± s.d. (c-d) Compound 1c (10-100 μM) increased NRF2 activity in the presence of pathological, but not physiological concentrations of (c) hydrogen peroxide (H2O2), (d) or peroxynitrite (ONOO−) in vitro. N = 3/group. Extra sum-of-squares F tests, not significant (n.s.), ***P < 0.001. Data are mean ± SD. (e) NRF2 activity induced by 1c per se is blocked by peroxide decomposition catalysts. N = 3/group. Extra sum-of-squares F test, ***P < 0.001. Data are mean ± s.d.
Extended Data Fig. 2 Hydrogen peroxide triggered monomethyl fumarate (MMF) release from compound 1c.
(a) Compound 1c (500 µM) was activated by 1.1 or 5 molar equivalents but not 0.01 molar equivalents of hydrogen peroxide (H2O2). Stacked HPLC traces of authentic samples of 1c and MMF, compared with reactions of 1c with H2O2 after 40 and 130 minutes in water. (b) Schematic of compound 1c reactivity in aqueous solutions and with H2O2. Compound 1c formed an equilibrium between the keto and hydrate forms in water and hydrolysed in pH 7.4 phosphate buffer to compound 1dwhich also formed an equilibrium between the keto and hydrate forms. The keto forms of compounds 1d and 1c can undergo Baeyer-Villi ger oxidation with H2O2 to release MMF. The hydrate forms of compounds 1d and 1c lack the 1,2-dicarbonyl moiety to react with H2O2. (c) Excerpt of the alkene region from 1H NMR spectra of MMF in pH 7.4 phosphate buffer, 1c in water and 1c in pH 7.4 phosphate buffer. MMF appeared as a single species, whilst 1c was present in the keto and hydrate forms in water. In pH 7.4 phosphate buffer, 1c (present in the keto and hydrate forms) hydrolysed to α-ketoester 1dalso in the keto and hydrate forms. (d) Excerpts of the alkene region and the 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS)-d6 peak from 1H NMR spectra showing the reactivity of compound 1c with H2O2 in pH 7.4 phosphate buffer. Compound 1c is converted cleanly and completely to MMF by 2 molar equivalents of H2O2 after 86.1 minutes. The presence of only MMF and 1c in the hydrate form suggests H2O2 reacts rapidly with the keto form of 1c upon conversion from the hydrate form, releasing MMF. 1H NMR spectra taken with an internal reference of 10 mM DSS-d6.
Extended Data Fig. 3 α-Ketoacid glutathione S-conjugate 4d is the major circulating species after oral 1c administration.
(a) Proposed metabolic profile of α-ketoester 1c. (b) Plasma concentrations of metabolites after a single oral dose of 1c (350 μmol/kg). N = 6 male rats. No data point shown if metabolite was not detected at the specific time. Data are mean ± s.d. Raw data in Supplementary Data Table 1.
Extended Data Fig. 4 Compound 1c preferentially activates NRF2 at sites of oxidative stress alive.
(a) In contrast to diroximel fumarate (DRF), 1c preferentially increased NRF2 target gene expression in L4/5 dorsal root ganglia (DRG) ipsilateral to peripheral nerve injury. Compound 1c and DRF were orally dosed at 350 μmol/kg/day. N = 3 mice/sex/group. Two-way ANOVA with Tukey’s post hoc tests, *P < 0.05, **P < 0.01, ***P < 0.001. Data are mean ± s.d. Individual replicates presented in grey dots. (b) Co-administration of catalase and FeTMPyP prevented expression of genes encoding antioxidants induced by 1c in L4/5 DRG ipsilateral to peripheral nerve injury. Compound 1c and DRF were orally dosed at 350 μmol/kg/day. N = 3 mice/sex/group. Unpaired, two-tailed T tests, **P < 0.01, ***P < 0.001. Data are mean ± s.d. Individual replicates presented in grey dots. (c) Glutathione S-conjugate 4d preferentially increased NRF2 nuclear translocation in ipsilateral L4/5 DRG, in contrast to monomethyl fumarate (MMF). Compound 4d and MMF were intravenously dosed at 190 μmol/kg/day. DRG from 2 mice pooled/sample; n = 2 samples/sex/group. Two-way ANOVA and Tukey’s post hoc test. *P < 0.05, **P < 0.01. (d) In contrast to DRF, 1c did not increase NRF2 nuclear translocation in heart, lung, liver, or kidney. Compound 1c and DRF were orally dosed at 350 μmol/kg/day. Ν=3 mice/sex/group. One-way ANOVA with Tukey’s post hoc tests, ***P < 0.001. Data are mean ± s.d. Individual replicates presented in grey dots.
Source data
Extended Data Fig. 5 Compound 1c relieves chronic pain alive.
(a) Oral 1c and diroximel fumarate (DRF) reversed dynamic allodynia 6 months after peripheral nerve injury. Compound 1c and DRF were orally dosed at 350 μmol/kg/day, represented by grey boxes. N = 4 mice/sex/group. Friedman with Dunn post hoc test, relative to 1c day 180: §§§P < 0.001; relative to DRF day 180: †P < 0.05, ††P < 0.01, †††P < 0.001. Data are median ± range. (b) Oral 1c and DRF reversed cold allodynia 6 months after peripheral nerve injury. Compound 1c and DRF were orally dosed at 350 μmol/kg/day, represented by grey boxes. N = 4 mice/sex/group. Repeated measures two-way ANOVA with Tukey’s post hoc tests, relative to vehicle: *P < 0.05, **P < 0.01, ***P < 0.001; 1c vs. DRF: #P < 0.05, ##P < 0.01, ###P < 0.001. Data are mean ± s.d. Individual replicates presented in spaghetti plots. (c) Compound 1c did not alter punctate allodynia in sham-operated animals after three days of treatment (unpaired, two-tailed t test). Compound 1c dose-dependently reversed punctate allodynia induced by peripheral nerve injury (PNI) (one-way ANOVA with Dunnett’s post hoc test). Relative to vehicle (0 μmol/kg): ***P < 0.001. Anti-allodynic effects of DRF are shown after equimolar treatment. N = 3 mice/sex/group. Data are mean ± s.d. (d) Compound 1c did not alter dynamic allodynia in sham-operated animals after three days of treatment (two-tailed Mann-Whitney test). Compound 1c dose-dependently reversed dynamic allodynia induced by peripheral nerve injury (Kruskal Wallis with Dunn’s post hoc test). Relative to vehicle (0 μmol/kg): *P < 0.05, ***P < 0.001. Anti-allodynic effects of DRF are shown after equimolar treatment. N = 3 mice/sex/group. Data are median ± range. (e) Five-day treatment with oral 1c (350 μmol/kg/day) did not result in loss of anti-allodynic efficacy after peripheral nerve injury, compared to subcutaneous morphine (3 mg/kg b.i.d.). N = 3 mice/sex/group. One-way ANOVA with Dunnett’s post hoc tests. Relative to 1c at day 0 (D0, prior to surgery): **P < 0.01, ***P < 0.001; relative to morphine D0: #P < 0.05, ###P < 0.001. Data are mean ± s.d. (f-g) There were no genotype differences in the effects of morphine (5 mg/kg, s.c.) on (f) punctate (repeated measures three-way ANOVA with Šídák’s post hoc test) or (g) dynamic allodynia (two-tailed Mann-Whitney test). N = 4 mice/sex/group. Relative to vehicle: ***P < 0.001. Data are mean ± s.d. for punctate allodynia and median ± range for dynamic allodynia. (h) Glutathione S-conjugate 4d and monomethyl fumarate (MMF) reversed punctate allodynia after peripheral nerve injury. Compound 4d and MMF were intravenously dosed at 190 μmol/kg/day. N = 4 mice/sex/group. Repeated measures two-way ANOVA with Tukey’s post hoc tests, relative to vehicle: ***P < 0.001. (i-j) Oral 1c attenuated (i) punctate allodynia and (j) numbness after two cycles of cisplatin (hatched boxes) were complete. Compound 1c was orally dosed at 350 μmol/kg/day, represented by grey box. N = 4 mice/sex/group. Repeated measures two-way ANOVA with Tukey’s post hoc test, or unpaired two-tailed t test. *P < 0.05, ***P < 0.001. Data are mean ± s.d. Individual replicates presented in grey dots. (k) Oral 1c attenuated punctate allodynia in mice chronically fed a high fat diet. Compound 1c was orally dosed at 225 μmol/kg/day, represented by grey box. N = 4 mice/sex/group. Repeated measures two-way ANOVA with Tukey’s post hoc test. *P < 0.05, ***P < 0.001. Data are mean ± s.d.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Reprints and permissions
About this article

Cite this article
Avery, T.D., Li, J., Turner, D.J.L. et al. Site-specific drug release of monomethyl fumarate to treat oxidative stress disorders. Nat Biotechnol (2024). https://doi.org/10.1038/s41587-024-02460-4
Download citation
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41587-024-02460-4







