
 Hot Deals
Hot Deals Shopfinish
Shopfinish Shop
Shop Appliances
Appliances Babies & Kids
Babies & Kids Best Selling
Best Selling Books
Books Consumer Electronics
Consumer Electronics Furniture
Furniture Home & Kitchen
Home & Kitchen Jewelry
Jewelry Luxury & Beauty
Luxury & Beauty Shoes
Shoes Training & Certifications
Training & Certifications Wears & Clothings
Wears & Clothings





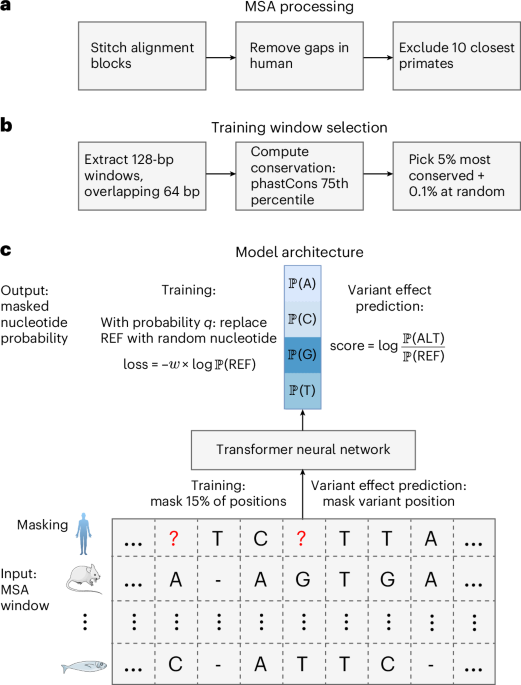
A DNA language model based on multispecies alignment predicts the effects of genome-wide variants
Goldfeder, R. L., Wall, D. P., Khoury, M. J., Ioannidis, J. P. & Ashley, E. A. Human genome sequencing at the population scale: a primer on high-throughput DNA sequencing and analysis. Am. J. Epidemiol. 1861000–1009 (2017).
Article PubMed PubMed Central Google Scholar
Marwaha, S., Knowles, J. W. & Ashley, E. A. A guide for the diagnosis of rare and undiagnosed disease: beyond the exome. Genome Med. 1423 (2022).
Lee, S., Abecasis, G. R., Boehnke, M. & Lin, X. Rare-variant association analysis: study designs and statistical tests. Am. J. Hum. Genet. 955–23 (2014).
Article CAS PubMed PubMed Central Google Scholar
Trajanoska, K. et al. From target discovery to clinical drug development with human genetics. Nature 620737–745 (2023).
Article CAS PubMed Google Scholar
Meier, J. et al. Language models enable zero-shot prediction of the effects of mutations on protein function. In Proc. Advances in Neural Information Processing Systems 34 (eds Ranzato, M. et al.) 29287–29303 (Curran Associates, Inc., 2021).
Brandes, N., Goldman, G., Wang, C. H., Ye, C. J. & Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 551512–1522 (2023).
Article CAS PubMed PubMed Central Google Scholar
Jagota, M. et al. Cross-protein transfer learning substantially improves disease variant prediction. Genome Biol. 24182 (2023).
Article CAS PubMed PubMed Central Google Scholar
Benegas, G., Batra, S. S. & Song, Y. S. DNA language models are powerful predictors of genome-wide variant effects. Proc. Natl Acad. Sci. USA 120e2311219120 (2023).
Article CAS PubMed PubMed Central Google Scholar
Dalla-Torre, H. et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat. Methods https://doi.org/10.1038/s41592-024-02523-z (2024).
Article PubMed Google Scholar
Vaswani, A. et al. Attention is all you need. In Proc. Advances in Neural Information Processing Systems 30 (eds Guyon, S. et al.) 6000–6010 (Curran Associates, Inc., 2017).
Armstrong, J., Fiddes, I. T., Diekhans, M. & Paten, B. Whole-genome alignment and comparative annotation. Annu. Rev. Anim. Biosci. 741–64 (2019).
Article CAS PubMed Google Scholar
Nguyen, E. et al. HyenaDNA: long-range genomic sequence modeling at single nucleotide resolution. In Proc. 37th International Conference on Neural Information Processing Systems (eds Oh, A. et al.) 43177–43201 (Curran Associates, Inc., 2023).
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 1331 (2021).
Article CAS PubMed PubMed Central Google Scholar
Pollard, K. S., Hubisz, M. J., Rosenbloom, K. R. & Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20110–121 (2010).
Article CAS PubMed PubMed Central Google Scholar
Sullivan, P. F. et al. Leveraging base-pair mammalian constraint to understand genetic variation and human disease. Science 380eabn2937 (2023).
Article CAS PubMed PubMed Central Google Scholar
Rives, A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl Acad. Sci. USA 118e2016239118 (2021).
Article CAS PubMed PubMed Central Google Scholar
Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 181196–1203 (2021).
Article CAS PubMed PubMed Central Google Scholar
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176535–548.e24 (2019).
Article CAS PubMed Google Scholar
Rao, R. M. et al. MSA Transformer. In Proceedings of the 38th International Conference on Machine Learning (eds Meila, M. & Zhang, T.) (PMLR, 2021).
Paten, B. et al. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 181829–1843 (2008).
Article CAS PubMed PubMed Central Google Scholar
Siepel, A. et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 151034–1050 (2005).
Article CAS PubMed PubMed Central Google Scholar
Landrum, M. J. et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 48D835–D844 (2020).
Article CAS PubMed Google Scholar
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 62592–100 (2024).
Article CAS PubMed Google Scholar
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47D886–D894 (2019).
Article CAS PubMed Google Scholar
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47D941–D947 (2019).
Article CAS PubMed Google Scholar
Notin, P. et al. ProteinGym: large-scale benchmarks for protein fitness prediction and design. In Proceedings of the Advances in Neural Information Processing Systems 37 (eds Oh, A. et al.) (NeurIPS, 2023).
Smedley, D. et al. A whole-genome analysis framework for effective identification of pathogenic regulatory variants in mendelian disease. Am. J. Hum. Genet. 99595–606 (2016).
Article CAS PubMed PubMed Central Google Scholar
Albuisson, J. et al. Identification of two novel mutations in Shh long-range regulator associated with familial pre-axial polydactyly. Clin. Genet. 79371–377 (2011).
Article CAS PubMed Google Scholar
Kvon, E. Z. et al. Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants. Cell 1801262–1271.e15 (2020).
Article CAS PubMed PubMed Central Google Scholar
Arbini, A. A., Pollak, E. S., Bayleran, J. K., High, K. A. & Bauer, K. A. Severe factor VII deficiency due to a mutation disrupting a hepatocyte nuclear factor 4 binding site in the factor VII promoter. Blood 89176–182 (1997).
Article CAS PubMed Google Scholar
Gao, H. et al. The landscape of tolerated genetic variation in humans and primates. Science 380eabn8153 (2023).
Article CAS PubMed PubMed Central Google Scholar
The Dependency Map Consortium. DepMap 23Q4 public. figshare https://doi.org/10.25452/figshare.plus.24667905.v2 (2023).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581434–443 (2020).
Article CAS PubMed PubMed Central Google Scholar
Agarwal, I., Fuller, Z. L., Myers, S. R. & Przeworski, M. Relating pathogenic loss-of-function mutations in humans to their evolutionary fitness costs. eLife 12e83172 (2023).
Article CAS PubMed PubMed Central Google Scholar
Zeng, T., Spence, J. P., Mostafavi, H. & Pritchard, J. K.Bayesian estimation of gene constraint from an evolutionary model with gene features. Nat. Genet. 561632–1643 (2024).
Article CAS PubMed Google Scholar
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. The gene. With. 17405–424 (2015).
Schneider, T. D. & Stephens, R. M. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 186097–6100 (1990).
Article CAS PubMed PubMed Central Google Scholar
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12996–1006 (2002).
Article CAS PubMed PubMed Central Google Scholar
Nair, S. et al. The dynseq browser track shows context-specific features at nucleotide resolution. Nat. Genet. 541581–1583 (2022).
Article CAS PubMed PubMed Central Google Scholar
Fishman, V. et al. GENA-LM: a family of open-source foundational models for long DNA sequences. Preprint at bioRxiv https://doi.org/10.1101/2023.06.12.544594 (2023).
Borgeaud, S. et al. Improving language models by retrieving from trillions of tokens. In Proc. 39th International Conference on Machine Learning (eds Chaudhuri, K. et al.) 2206–2240 (PMLR, 2022).
Weiner, D. J. et al. Polygenic architecture of rare coding variation across 394,783 exomes. Nature 614492–499 (2023).
Article CAS PubMed PubMed Central Google Scholar
Weissbrod, O. et al. Functionally informed fine-mapping and polygenic localization of complex trait heritability. Nat. Genet. 521355–1363 (2020).
Article CAS PubMed PubMed Central Google Scholar
Márquez-Luna, C. et al. Incorporating functional priors improves polygenic prediction accuracy in UK Biobank and 23andMe data sets. Nat. Common. 126052 (2021).
Article PubMed PubMed Central Google Scholar
Aw, A. J., McRae, J., Rahmani, E. & Song, Y. S. Highly parameterized polygenic scores tend to overfit to population stratification via random effects. Preprint at bioRxiv https://doi.org/10.1101/2024.01.27.577589 (2024).
Blanchette, M. et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14708–715 (2004).
Article CAS PubMed PubMed Central Google Scholar
Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (eds Burstein, J. et al.) 4171–4186 (Association for Computational Linguistics, 2018).
Su, J. et al. RoFormer: enhanced transformer with rotary position embedding. Neurocomputing 568127063 (2024).
Article Google Scholar
Wolf, T. et al. Transformers: state-of-the-art natural language processing. In Proc. 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations (eds Liu, Q. & Schlangen, D.) 38–45 (Association for Computational Linguistics, 2020).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17122 (2016).
Article PubMed PubMed Central Google Scholar
Bolognesi, B. et al. The mutational landscape of a prion-like domain. Nat. Common. 104162 (2019).
Article PubMed PubMed Central Google Scholar
Consortium, G. P. et al. A global reference for human genetic variation. Nature 52668–74 (2015).
Article Google Scholar
Zhou, H. et al. FAVOR: functional annotation of variants online resource and annotator for variation across the human genome. Nucleic Acids Res. 51D1300–D1311 (2023).
Article PubMed Google Scholar
McVicker, G., Gordon, D., Davis, C. & Green, P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 5e1000471 (2009).
Article PubMed PubMed Central Google Scholar
Gazal, S. et al. Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet. 491421–1427 (2017).
Article CAS PubMed PubMed Central Google Scholar
Benegas , G. , Albors , C. , Aw , AJ , Ye , C. & Song , YS GPN repository . GitHub https://github.com/songlab-cal/gpn (2024).
Discover more from Tamfis Nigeria Lmited
Subscribe to get the latest posts sent to your email.











