
 Hot Deals
Hot Deals Shopfinish
Shopfinish Shop
Shop Appliances
Appliances Babies & Kids
Babies & Kids Best Selling
Best Selling Books
Books Consumer Electronics
Consumer Electronics Furniture
Furniture Home & Kitchen
Home & Kitchen Jewelry
Jewelry Luxury & Beauty
Luxury & Beauty Shoes
Shoes Training & Certifications
Training & Certifications Wears & Clothings
Wears & Clothings





Precise in vivo RNA base editing with a wobble-enhanced circular CLUSTER guide RNA
Technology tamfitronics
Main
Site-directed adenosine-to-inosine (A-to-I) RNA base editing is a very promising technology with a clear path for clinical application1,2. Hydrolytic deamination of A by enzymes of the ADAR family (adenosine deaminases acting on RNA) produces an inosine, which is biochemically interpreted as G in many cellular processes such as splicing or translation and, consequently, functionally substitutes A with G on the RNA level3,4. There are three catalytically active human ADAR proteins: constitutively and ubiquitously expressed ADAR1 p110, interferon-inducible ADAR1 p150 and ADAR2. In the past, ADAR deaminase domains have been engineered into various artificial editing approaches that enable the efficient and highly programmable editing of any given target A in the transcriptome by applying customized guide RNAs and simple Watson–Crick base-pairing rules. Typical examples are the SNAP-ADAR5,6the λN-ADAR7,8,9 and the Cas13-ADAR approaches10,11,12. However, even after several rounds of optimization, major limitations of such systems remain: (1) a guide RNA plus a protein component needs to be delivered; (2) nonhuman protein domains are included; and (3) global off-target editing hampers the clinical development. An elegant solution to all three limitations could be to harness ubiquitously expressed endogenous ADAR enzymes for RNA base editing. It was shown recently that endogenous ADAR1 can be recruited by either chemically modified antisense oligonucleotides (ASOs)13,14 or genetically encoded guide RNAs15,16,17,18. Genetically encoded guide RNAs are particularly desired for the long-lasting reversal of disease-causing G>A point mutations in vivo by viral delivery of the guide RNA component.
Genetically encoded guide RNAs currently suffer from massive bystander editing in the guide RNA–target RNA duplex. This is, on one hand, caused by the large size of the duplex (70–200 bp) and, on the other hand, by the ubiquitous presence of highly editable A bases all over the duplex. Both ADAR1 and ADAR2 prefer similar nearest neighboring bases (for example, U > A > C > G at the 5′ position relative to the target A and G > C ≈ A > U or G > C > U ≈ A at the 3′ position, respectively)19. Consequently, bystander editing is dominated by a handful of preferred triplets, particularly all four 5′-UAN triplets (N = A, U, G or C), 5′-AAG and 5′-CAG. Bystander editing can lead to unwanted recoding events in the target and might even cause ribosome stalling20. Thus, the avoidance of bystander editing represents a major engineering problem for encodable RNA base-editing systems. Today, several strategies have been suggested. In the LEAPER approach16,17bystander editing is usually suppressed by mismatching some or even all bystander-prone A bases with G. The rationale behind this is the preference of the ADAR deaminase for a specific counter base (C > U > A or G) opposite the targeted A21. A more recent approach is U depletion of the guide RNA by keeping off-target prone A bases unpaired, thus producing single-nucleotide bulges within the guide RNA–mRNA duplex20. However, both strategies do not always work optimally as they sometimes fail to suppress bystander editing and often reduce editing efficiency, sometimes even dramatically (for example, in A-rich sequence contexts). Consequently, using these strategies to improve editing precision can cost notable editing efficiency. An alternative solution is presented in the CLUSTER approach15. Here, the presence of editable triplets in the guide RNA–mRNA duplex is minimized by subdividing the guide RNA into several functional segments. Each segment is designed to bind the target transcript in areas selected for the absence of editable A bases. Because the individual segments of the CLUSTER guide RNAs, which we refer to as recruitment sequences (RSs), can be arranged closely within a window of a few hundred nucleotides around the target site, the approach leads to a very good control of bystander editing and high editing yields. However, in target transcripts that are very rich in highly editable A bases, the RSs currently have to be placed at large distances, which sometimes compromises editing efficiency. In a somewhat related solution, the guide RNA–mRNA duplex is regularly interrupted by bulged-out nucleotides, which leads to a reduction in bystander editing18. While the latter guide RNAs are very simple to design, the CLUSTER approach allows the assembly of guide RNAs from a much larger sequence space and was shown to enable a boost of editing efficiency by computationally selecting CLUSTER guide RNAs that avoid inhibitory self-folding.
In the human transcriptome, several highly efficient and precise editing events are known, which are guided by cis-acting intronic editing complementary sequences (ECS) that fold back to the target site inside an exon22,23,24,25,26,27,28. Notably, such natural editing sites are typically not found in perfect RNA helices but rather contain bulges, mismatches and wobble base pairs, which may serve to suppress bystander editing while preserving high on-target editing yields. The exact structural layer that makes an RNA a good or poor substrate for ADAR is still underexplored, although several recent studies have started to address this issue29,30,31.
Here, we systematically explore the rational use of G•U wobble base pairs to affect editing precision and efficiency. The G•U wobble base pair is the most abundant type of non-Watson–Crick base pair in the transcriptome32,33. Surrounding the G•U wobble base, the RNA helix structure is perturbed, affecting the groove width, base stacking and electrostatic profile33. Specific structural effects induced by G•U wobble bases have been shown to be important for the interaction of double-stranded RNA (dsRNA)-binding proteins with dsRNA substrates, including ADAR34. In this study, we systematically deduce rules on how to place G•U wobble base pairs to improve site-directed RNA base editing in terms of editing precision and efficiency. We apply G•U wobble base pairs in various RNA base-editing approaches including the CLUSTER approach, where it greatly improves the guide RNA design process, giving raise to highly precise and efficient circular CLUSTER guide RNAs that recode a Rett syndrome causing mutation in vivo. The disease in the model animals is caused by a loss-of-function mutation (W104>amber) in the transcription factor methyl CpG-binding protein 2 (MeCP2) that impairs its expression and binding to methylated DNA, thus deregulating>2,500 downstream genes, including many that are relevant for neuronal function35. This model system allowed us to assess key factors of successful in vivo transcript repair with endogenous Adars in the central nervous system (CNS).
Results and discussion
Wobble bases modulate editing depending on their orientation
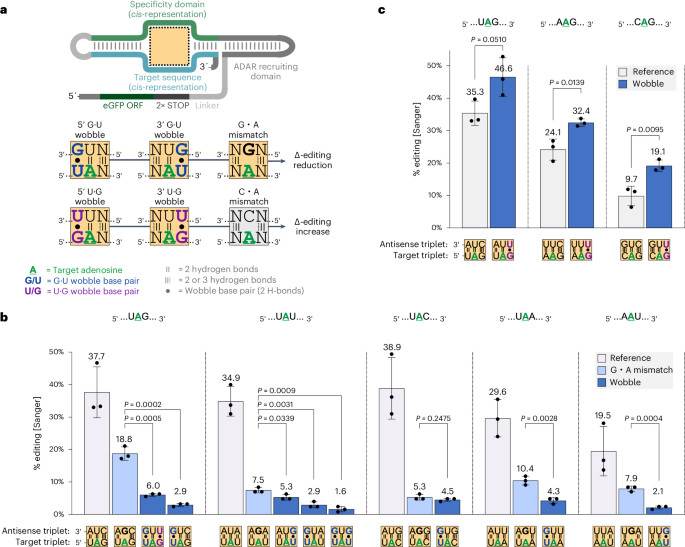
To characterize the effect of wobble base pairs in the nearest neighbor context, we first studied all 12 possible triplets that contained either a U or a G either 5′ and/or 3′ next to the target A. While the target A was always placed opposite of a U base, similar to a regular bystander site, the nearest neighboring nucleotides were either conventionally Watson–Crick base-paired or wobble base-paired (Fig. 1a). Under these circumstances, four types of wobble base pairs can occur. When the U base in a 5′-UAN or 5′-NAU triplet (N = any base) is base-paired with a G, we refer to this as a 5′-G•U or 3′-G•U wobble, respectively; when the G base in a 5′-GAN or 5′-NAG triplet is base-paired with a U, we refer to this as a 5′-U•G or 3′-U•G wobble, respectively. As a benchmark to previous studies21we also included experiments where the target A was mismatched with a G base (G•A mismatch), to suppress editing.

aDesign of the cis-acting editing reporter and illustration of the applied triplet base-pairing motifs. The orientation of wobble base pairs at nearest neighbor positions or the presence of G or C counter bases modulate Δ-editing at the central A of a triplet in comparison to its fully Watson–Crick base-paired counterpart. All triplet base-pairing motifs with ocher background color were installed at the empty dotted outline within the cis-acting editing construct to generate the results in b,c. bSuppression of RNA editing in different target triplets using G•U wobble base pairs or G•A mismatches. cEnhancing RNA editing in different target triplets using U•G wobble base pairs. The Sanger sequence analysis in b and c was performed after transfection of editing reporter plasmids into ADAR1 p110 Flp-In T-REx cells. Data in b and c are shown as the mean ± s.d. of n = 3 biological replicates. For statistical analysis, a Student’s t-test (two-tailed, parametric) was applied.
The experiment used an editing reporter construct, based on the earlier R/G-guide RNA approach36,37. While the trans-acting guide RNA comprised only a double-stranded ADAR-recruiting domain and a single-stranded 20-nt specificity domain (SD), the cis-acting reporter additionally contained its own 20-nt target sequence (TS) and was located in the 3′ untranslated region (UTR) of an eGFP transcript to enable convenient Sanger sequencing readout (Fig. 1a). Editing was performed by transfecting the plasmid-borne editing reporter into Flp-In T-REx cells, overexpressing ADAR1 p110.
Importantly, we found that both, 5′-G•U and 3′-G•U wobble base pairs strongly suppress editing in the five triplets, 5′-UAN and 5′-AAU (Fig. 1b and Supplementary Fig. 1), which are highly editable under normal Watson–Crick base-pairing conditions and are, thus, a major source of bystander off-target events for trans-acting guide RNAs. In the 5′-UAU triplet, both G•U wobbles seemed to cooperate. For four of the five triplet contexts, the suppressive effect of the G•U wobble on editing significantly outcompeted the suppressive effect of the G•A mismatch (Fig. 1b). Interestingly, we found the opposite effect for the U•G wobble base pair. In particular, for the three triplets 5′-UAG, 5′-AAG and 5′-CAG, a clear editing-enhancing effect on the A directly adjacent to the 3′-U•G wobble was apparent (Fig. 1 C). Because of the inability of ADARs to achieve sufficient editin g at 5′-GAN triplets, the effect of 5′-U•G wobbles could not be verified (Supplementary Fig. 1).
The 5′-UAG triplet can simultaneously accommodate both a suppressive 5′-G•U and an enhancing 3′-U•G wobble base pair and, thus, allows studying their interplay. Our data suggest that the suppressive effect of the G•U wobble entirely dominates the activating effect of the U•G wobble (Fig. 1b).
It is intriguing to speculate that the enhancing effect of the 3′-U•G wobble could be combined with or replace the activating effect of the commonly used C•A mismatch at an on-target site such as 5′-UAG. We tested this idea using the CLUSTER guide RNA system on three endogenous targets (Supplementary Fig. 2a,b), with a mixed outcome. Only in one example (ACTB) was the U•G wobble a promising alternative to the C•A mismatch, although it could be useful in certain other sequence contexts for guide RNA structure optimization.
A guide RNA might find and bind to near-cognate sequences within the transcriptome to induce off-target editing. In this context, we aimed to understand whether a randomly occurring G•U wobble suppresses editing only in the context of a U-paired A or also in the rare context of a C-mismatched A, which is known to be more prone to editing. We evaluated the potential of 5′-G•U wobbles for suppressing editing at 5′-UAG sites where the A was mismatched with C. In two of three examples (GUSB and NUP43), the suppressive effect of the 5′-G•U wobble was strong enough to suppress editing even at the mismatched A (Supplementary Fig. 2c).
To see whether the enhancing and suppressing effects of wobble base pairs also apply for ADAR1 p150 and ADAR2, we selected the 5′-UAG triplet, which is a particularly frequent site of bystander editing, and transfected the corresponding plasmid-borne editing reporters into Flp-In T-REx cells, overexpressing ADAR1 p150 or ADAR2, respectively. As expected, the underlying mechanisms were not ADAR isoform dependent and worked equally well (Extended Data Fig. 1).
G•U wobbles improve precision and efficiency of LEAPER guides
We next applied G•U wobble base pairs in the context of unstructured 111-nt-long LEAPER guide RNAs16 (Fig. 2a and Supplementary Fig. 3), which are highly prone to bystander editing. Our data (Fig. 1b) indicated that G•U wobble base pairs would be convenient to suppress bystander editing in the five highly editable triplets 5′-UAN (N = A, U, G or C) and 5′-AAU and could be combined with the G•A mismatch at all other editable triplets, such as 5′-AAG and 5′-CAG. To test this concept, trans-acting LEAPER guide RNAs (Fig. 2a) encoded on plasmids were cotransfected into HeLa cells with plasmids carrying the full-length complementary DNA (cDNA) of one of three different target genes (AHI1 (ref. 38), COL3A1 (ref. 39) and BMPR2 (ref. 40)) each carrying a disease-relevant W>amber STOP mutation (5′-UAG) (Fig. 2b–d). To also evaluate endogenous targets, LEAPER guide RNAs targeting a 5′-UAG within the 3′ UTR of NUP43 and RAB7A were transfected into HEK293FT cells expressing these genes (Extended Data Fig. 2a,b). As seen in both settings, LEAPER guide RNAs recruited endogenous ADAR to induce significant on-target editing in each of the five targets (54–80%); however, this was contaminated with massive bystander editing (>10 sites per target), as previously reported15.

aSchematic of 111-nt-long unstructured linear LEAPER guide RNA. b–dEditing heat maps of the LEAPER guide RNA-binding sites within the indicated transcripts: AHI1 (b), COL3A1 (c) and BMPR2 (d). The basic design column, LEAPER, lacks G•A mismatches and wobble base pairs. The other guide RNAs contain either G•A mismatches or G•U wobbles at G•U-amenable sites or a combination of both solutions at all bystander sites. In the latter case, G•A mismatches are placed at sites not amenable to G•U wobbles. The triplet context for each listed editing event is given with the target A highlighted in green and all off-target A bases in blue. The position of each site is given relative to the transcript and the target A (±0 position). Editing was performed with plasmid-borne guide RNA and target in HeLa cells (endogenous ADAR). Data are shown as the mean editing percentage ± s.d. of n = 3 (AHI1 and COL3A1) or n = 5 (BMPR2) biological replicates.
First, we tested for all five targets whether G•U wobble base pairs outcompete G•A mismatches to suppress bystander editing at such sites, where G•U wobbles are amenable (G•U at G•U-amenable sites) and compared such guide RNAs with guide RNAs that apply G•A mismatches at the same sites (G•A at G•U-amenable sites) (Fig. 2b–d and Extended Data Fig. 2a,b). Notably, we found that the G•U wobble strategy was very potent to suppress bystander editing, often but not always outcompeting the G•A mismatch approach. Moreover, the on-target editing yield was higher for four of the five targets (AHI174% versus 51%, and COL3A147% versus 29%, Fig. 2; endogenous NUP4336% versus 17%, and endogenous RAB7A45% versus 25%, Extended Data Fig. 2).
Second, we aimed to suppress bystander editing entirely by either fully relying on the prior-art G•A mismatch approach (G•A all off-target sites) or by combining G•A mismatches with G•U wobble base pairs (G•A and G•U at all off-target sites). For AHI1 (Fig. 2b), only the combination of G•A mismatches with G•U wobble base pairs achieved the entire suppression of bystander editing and a good on-target editing yield (45%), while the guide RNA using only G•A mismatches suffered from residual bystander editing (position −38, 43%; position −35, 15%) and a reduced on-target efficiency (26%). These results highlight the power of G•U wobbles to improve the precision of LEAPER guide RNAs. For COL3A1 (Fig. 2c) and NUP43 (Extended Data Fig. 2a), the on-target yields were again clearly better when complementing G•A mismatches with wobble base pairs (41% versus 21% and 23% versus 12%, respectively). However, because of the lower yields at all edited sites, the G•A mismatch approach appeared to give slightly better precision. The BMPR2 target transcript (Fig. 2d) gave equal on-target yields for both strategies but better precision when G•U wobble base pairs were included. In the case of RAB7A (Extended Data Fig. 2b), both designs performed similarly.
According to the literature, LEAPER guide RNAs that carry numerous G•A mismatches can suffer from a loss of editing efficiency16. The reason for this might be that G•A mismatches have the lowest duplex stability among all known nucleotide mismatches41. In contrast, the thermodynamic stability of the G•U wobble is comparable to the A-U base pair32. This might partly explain why on-target efficiency often benefitted when G•A mismatches were complemented with G•U wobble base pairs. Notably, the number of suppressive G•U wobble base pairs required to improve precision can be smaller than that of G•A mismatches because one G•U wobble acts simultaneously at its 5′ and 3′ nearest neighbor positions. Examples can be found in Fig. 2b,c (AHI1positions +29 and +31; COL3A1positions −5 and −3). Furthermore, we found several cases where G•A mismatching failed to fully suppress bystander editing, while the G•U wobble strategy succeeded (for example, AHI1 at positions −38, −35 and −31 and BMPR2 at positions −32, +32 and +35; Figure 2b,d).
After targeting both exogenous and endogenous transcripts ranging from high expression levels (AHI1, BMPR2 and COL3A1 all as cDNAs), over medium (RAB7Anormalized transcripts per million (nTPM) = 105) to low (NUP43nTPM = 36) expression levels, our data suggest that the G•U wobble strategy is unaffected by target transcript abundance and, thus, widely applicable. Overall, the wobble strategy complements the prior-art G•A mismatch strategy very well and regularly improves both editing efficiency and precision.
G•U wobbles are widely applicable to RNA base-editing tools
Bystander editing is a common problem of all RNA base-editing systems, particularly those such as the λN-ADAR8 and Cas13-ADAR10 approaches (Fig. 3a–d and Supplementary Fig. 3) that also use genetically encoded guide RNAs. Thus, we applied the G•U wobble strategy with boxB guide RNAs (Fig. 3a) and direct repeat (DR) guide RNAs (Fig. 3c) to see whether it improves the precision of the λN-ADAR and the Cas13-ADAR approaches, respectively. For this, triple-plasmid protocols were used where plasmids encoding the guide RNA, the editase (λN-ADAR2Q or Cas13-ADAR2Q) and the target (AHI1 W725X) were cotransfected into HeLa cells. In contrast to the LEAPER approach, the λN-ADAR and Cas13-ADAR approaches apply a hyperactive ADAR mutant and use guide RNAs with comparably short antisense part (boxB, 49 nt; DR, 59 nt). Because of the shorter duplex, the number of bystander sites is overall smaller. Nevertheless, the λN-ADAR approach induced significant bystander editing (Fig. 3b). Notably, amenable sites were readily controlled by G•U wobbles but not by G•A mismatches alone. A combination of G•U wobble and G•A mismatches was able to fully suppress bystander editing at a minor cost of editing efficiency (65% ± 7% to 49% ± 2%; Fig. 3b). For Cas13-ADAR, the combination of G•U wobble and G•A mismatches gave the best results in terms of editing efficiency (25% ± 3% versus 20% ± 4%) and allowed for complete bystander suppression (Fig. 3d). However, the Cas13-ADAR system itself gave dramatically lower editing yields compared to the λN-ADAR system (26% ± 2% versus 65% ± 7%) and showed little specificity for the Cas13b protein, as the editing yield of the DR guide RNA with and without overexpression of the editase differed only by ~9% (Fig. 3d).

aSchematic of the λN-ADAR editing system. The 2× boxB motif-containing guide RNA (84 nt) binds its target mRNA (through 49 bp) to recruit the engineered hyperactive λN-ADAR2Q editase. bEditing heat map of exogenous AHI1 W725>amber mRNA targeted by the λN-ADAR system. cSchematic of the Cas13b-ADAR system. The DR motif-containing guide RNA (87 nt) binds its target mRNA (through 51 bp) to recruit the engineered PspCas13b-ADAR editase carrying a hyperactive ADAR2 E488Q deaminase domain. dEditing heat map of exogenous AHI1 W725>amber mRNA targeted by the Cas13b-ADAR system. eSchematic of a symmetric ASO bound to its target mRNA (through 58 nt). The ASO is end-blocked by 2′-OMe, contains phosphorothioate linkages and recruits endogenous ADARs. fEditing heat map of the ASO binding site within exogenous PEX1G843D transcript. In b,d,fthe basic designs (columns 2× boxB, DR guide RNA and ASO) do not contain G•A mismatches, wobble base pairs or 2′-OMe modifications beyond the end-blocks. The other guide RNAs contain additional 2′-OMe modifications, G•A mismatches, G•U wobbles at G•U-amenable sites or a combination of these solutions at all bystander sites. In the case of a combined solution, G•A mismatches or 2′-OMe modifications are placed at sites not amenable to G•U wobbles. The triplet context for each listed editing event is given with the target A highlighted in green and all off-target A bases in blue. The position of each site is given relative to the transcript and the target A (±0 position). Editing was performed in HeLa cells using plasmid-borne guide RNAs (2× boxB and DR guide RNA) and editase (λN-ADAR2Q and Cas13b-ADAR2Q) or ASOs recruiting endogenous ADAR. Data are shown as the mean editing percentage ± s.d. of n = 3 biological replicates.
In ADAR-recruiting ASOs, the strategic placement of chemical modifications allows to control bystander events13,14. However, dense chemical modification, for example with 2′-O-methylated ribose (2′-OMe), can interfere strongly with editing. Thus, we evaluated G•U wobble base pairs in a case where additional chemical modifications diminished the on-target efficiency. Using a chemically modified (phosphorothioate linkage and 2′-OMe end-blocked) 59-nt-long symmetric ASO (Fig. 3e), we targeted the PEX1 transcript, specifically the G843D substitution causative for the peroxisome biogenesis disorder Zellweger syndrome42. While placement of additional 2′-OMe modifications at the −25, −6 and +7 positions controlled bystander editing, they also reduced the on-target yield drastically (34% ± 7% to 12% ± 3%; Fig. 3f). By contrast, a combination of G•U wobble base pairs and 2′-OMe modifications enabled the control of bystander editing while preserving the on-target yield (28% ± 8%; Fig. 3f). The latter example shows that ASO-based approaches can also potentially benefit from wobble base pairs to maintain high on-target yields.
Superior off-target control in A-rich target sites
The suppression of bystander editing in closest proximity to an on-target A is a common problem for all fully encoded RNA base-editing systems. Strategies such as G•A mismatching16 or U depletion17 often lead to a substantial loss of editing yield when they are applied too close to the on-target site. To assess the G•U wobble strategy for such a setting, we systematically tested how far the suppressive effect of a single G•U wobble base pair extends in the 5′ and 3′ directions. For this purpose, we again used cis-acting constructs that placed an editable duplex in direct extension of an ADAR-recruiting domain (R/G helix; Extended Data Fig. 3a and Supplementary Fig. 4) into the 3′ UTR of an eGFP reporter. We then evaluated the editing yields after transfection of these constructs into ADAR1 p110-expressing Flp-In T-Rex 293 cells (Supplementary Fig. 4). To study the suppressive effect of the 3′-G•U wobble in the 5′ direction, we studied a series of three 5′-UA(A)iU base-paring motifs (i = 1–3 A bases) with increasing distance between the 3′-G•U wobble and the on-target A (Supplementary Fig. 4a–c). Accordingly, we also designed a series of three 5′-U(A)iAG base-paring motifs (i = 1–3 A bases) to test the effect of a 5′-G•U wobble in the 3′ direction (Supplementary Fig. 4d–f). The target triplet was either 5′-UAA or 5′-AAG, hereinafter indicated by square brackets. In both series, we also benchmarked the effect of the G•A mismatch for the same bystander off-target A site. Neither the 5′-G•U nor the 3′-G•U wobble affected the on-target editing yield negatively at any distance tested. Instead, the suppressive effect was almost entirely focused on the direct 3′ and 5′ neighboring base. This was in clear contrast to the G•A mismatch where not only the mismatched base but also the first and sometimes even the second neighboring base in both directions were negatively affected (Supplementary Fig. 4). Thus, the G•U wobble strategy should be particularly strong to precisely suppress bystander editing close to an on-target A. We show exemplary data on how the 5′-G•U wobble controls editing precision in a 5′-U[A[AAG]base-pairing motif (Extended Data Fig. 3a,c,e) and how the 3′-G•U wobble acts in the 5′-[U[UAA]U base-pairing motif (Extended Data Fig. 3a,d,f), always in comparison to the G•A mismatch. In both cases, the G•U wobble clearly gave a better balance of editing efficiency over editing precision.
Next, we transferred the concept to trans-acting CLUSTER guide RNAs (linear design), which targeted a 5′-U[A[AAG]site in the BMPR2 transcript (K984, on cDNA) by harnessing endogenous ADAR in HeLa cells (Extended Data Fig. 3b,c,g). As expected, the reference guide RNA showed good on-target yields (A59%; Extended Data Fig. 3g) but also a strong bystander editing at the 5′ neighboring A (30%). The strategically placed 5′-G•U wobble base pair was able to fully suppress this bystander editing. This was not the case with the G•A mismatch where only a partial suppression of bystander editing was achieved. Notably, the U depletion strategy17 even increased bystander editing to 38% (Extended Data Fig. 3g). The 5′-G•U wobble also gave the best on-target efficiency of the compared bystander solutions with 47% yield, whereas a G•A mismatch reduced the yield and U depletion almost fully blocked editing, highlighting the power of the G•U wobble strategy to achieve high efficiency and high precision in very A-rich triplet contexts where G•A mismatch and U depletion fail (Extended Data Fig. 3g and Supplementary Fig. 5a,b).
To show that this finding is highly generalizable, we performed a meta-analysis over three different target transcripts (AHI1, BMPR2 and COL3A1) representing three different A-rich target triplets (5′-[U[UAA]U, 5′-U[A[AAG]and 5′-U[A[AAA]U) while using two different editing approaches, the CLUSTER guide RNA with endogenous ADAR and the boxB/λN-ADAR system with engineered ADAR. Notably, not only bystander control but also on-target efficiency was significantly better with G•U wobble base pairs, demonstrating the strength of the strategy to suppress bystander editing precisely within A-rich triplets (Extended Data Fig. 3h and Supplementary Fig. 5).
Wobble bases improve the engineering of CLUSTER guide RNAs
CLUSTER guide RNAs represent a recent strategy to harness endogenous ADAR for precise and efficient RNA base editing15. The basic concept combines an ADAR recruitment motif, a 20-nt SD that binds the target site and three or more additional RSs 15–20 nt in length that bind to the target mRNA over a larger stretch of sequence space in a multivalent fashion (Supplementary Fig. 3). In silico optimization of the guide RNA sequence is applied to choose RSs in such a way that highly editable A bases are avoided, enabling high control over bystander editing. Furthermore, guide RNAs with a high tendency to form inhibitory secondary structure are sorted out automatically, which helps to improve editing efficiency. Similar to the report for the LEAPER system17we herein established the ribozyme-based Tornado expression system43 for circularization and, thus, stabilization of guide RNAs (Extended Data Fig. 4). Starting with a simple LEAPER design, we could verify the formation of cleanly circularized guide RNAs (Supplementary Fig. 6), which gave an improved editing efficiency on the endogenous RAB7A transcript (Supplementary Fig. 7). Particularly notable was the positive effect of circularization on the editing yield after stable integration of the LEAPER guide RNA cassette by the PiggyBac transposase into the genome of HeLa cells, which gave moderate and stable editing yields over several weeks even with the weaker Pol2 promoter, elongation factor 1α (EF1α) (Supplementary Fig. 8).
Compared to simple LEAPER guide RNAs, the flexible design of CLUSTER guide RNAs represented a considerable engineering challenge for circularization. The order of elements within the CLUSTER guide RNA, such as the SD, the RSs, the ADAR recruitment motif and the target mRNA exit points, can all be placed individually and relative to each other. On the guide RNA side, RSs can be placed 5′ and/or 3′ to the SD, while the ADAR recruitment motif can be flexibly placed anywhere in between. On the mRNA side, binding sites that correspond to the RS within the guide RNA can be located 5′ and/or 3′ of the TS, which corresponds to the SD within the guide RNA. Binding-site placement ultimately defines whether the exit points of the mRNA are close or distant relative to the ligation stem. Furthermore, the number and length of all antisense elements and linkers can be varied, which may individually affect the torsion within the guide RNA circle and, thus, its interaction with the target transcript.
To identify general design rules for circular CLUSTER guide RNAs, we tested various constructs on a luciferase reporter construct (Supplementary Figs. 9 and 10). We found that the mRNA exits should be placed approximately opposite of the SD and that the ADAR recruitment motif can be combined with the ligation stem into a larger RNA structure placed adjacent to the SD. Notably, a 5-nt bulge that separates ADAR recruitment motif and ligation stem achieved particularly high editing efficiency (Supplementary Fig. 10a–cdesign L13). This ADAR recruitment motif was called the split-R/G motif and was used in subsequent designs.
After identifying general design principles for circular CLUSTER guide RNAs, we aimed to design an optimal guide RNA for a proof-of-concept in vivo study. Specifically, we aimed to target a premature STOP codon in the murine Mecp2 transcript (W10 4>amber), which causes severe Rett syndrome-like symptoms in mice carrying this patient mutation44.
CLUSTER guide RNAs avoid bystander editing by choosing RSs that minimize the presence of editable A bases in the guide RNA–target RNA duplex. However, in highly A-rich target RNAs, such as Mecp2individual binding regions for RSs can be separated by long distances within the target transcript (for example, spread over several exons) and the available sequence space for secondary structure optimization can be limited. Both effects can negatively impact the editing efficiency of CLUSTER guide RNAs15. However, by applying the wobble base-pairing strategy to suppress bystander editing, the highly limiting filter set that defined eligible A bases within the RS-binding regions could now be considerably expanded; 5′-UAB, 5′-BAU triplets (B = C, G or U) and 5′-KAAU (K = G or U) sequence motifs are now included, while only 5′-GAB triplets were previously allowed15. Furthermore, 5′-CAC triplets and all A bases located at either end of a binding region (edge A bases), which we identified as being resistant to off-target editing, could be used to expand the sequence space. With the old filter settings (Fig. 4a), a circular CLUSTER guide RNA with four RSs and a split-R/G ADAR recruitment motif needs substantial space on the Mecp2 target RNA—specifically, 1,470 nt for guide RNA V1 and 1,208 nt for guide RNA V2 (Fig. 4c). In contrast, with the new filter settings (Fig. 4b), the entire guide RNA (V3) covers only 127 nt on the murine Mecp2 transcript (Fig. 4c). This guide RNA V3 gave significantly better on-target editing than V1 and V2 (87% versus 63% and 65%), which used the old filter settings (Fig. 4d). Partially, this might also be attributed to the larger available sequence space, which allows selecting guide RNAs with a much lower level of inhibitory secondary structure (Fig. 4e). As expected, the expansion of eligible A bases led to the appearance of bystander editing at binding sites of the RSs, which were albeit well suppressed with the G•U wobble strategy (Fig. 5).

aResults from an in silico search for RS-binding sites within the murine Mecp2 W104>amber ORF when applying the previous filter settings to the GuideRNA-Forge tool. The binding site for the SD is displayed in turquoise and outlined in black. It contains the target A in green and underlined. All potential RS-binding regions ≥ 20 nt length, in which binding sites for RSs can be selected, are displayed in light blue. The previous filter excludes all A bases except for those within a 5′-GAB (B = U, C or G) triplet context. bResults for the same in silico search with the latest filter that included A bases in a 5′-GAB, 5′-UAB, 5′-BAU, 5′-KAAU and 5′-CAC triplet context, in addition to allowing A bases independent of their sequence context at the edges of a detected binding region. cIllustration of binding regions in which specific binding sites of RSs of three circular CLUSTER guide RNAs are located within the Mecp2 ORF, generated using the previous filter (V1 and V2) or the latest filter (V3). dEditing was performed with plasmid-borne guide RNA and murine Mecp2 transcript in HeLa cells (endogenous ADAR). eSecondary structure prediction of guide RNA V1–V3 generated using the ViennaRNA Package 2.0 (ref. 54). The mean free energy (MFE) of the antisense part (SD and CLUSTER of RS) is given in kcal per mol. The dot–bracket ratio (DBR) indicates the number of dots (unpaired bases) per bracket (paired bases) within the dot–bracket annotation of the antisense part. Data are shown as the mean editing percentage ± s.d. of n = 3 biological replicates. For statistical analysis, a Student’s t-test (two-tailed, parametric) was applied.

Editing heat map of guide RNA-binding sites within the murine Mecp2 W104>amber transcript. The triplet context for each listed editing event is given with the target A highlighted in green and all off-target A bases in blue. The position of each site is given relative to the transcript and the target A (±0 position). Editing was performed with plasmid-borne guide RNA and murine Mecp2 transcript in HeLa cells (endogenous ADAR). aCircular CLUSTER guide RNA design. bCircular LEAPER guide RNA design. cYields achieved using circular unstructured LEAPER guide RNAs containing a 111-nt-long antisense part. The basic design (column circular LEAPER) does not contain bystander solutions. The other guide RNAs either contain G•A mismatches or apply U depletion at all bystander sites, G•U wobbles at G•U-amenable sites or a combination of G•A mismatching or U depletion with G•U wobbles at amenable sites. dYields achieved using circular CLUSTER guide RNAs containing a 100-nt-long antisense part split into a targeting sequence (20 nt) and four RSs (each 20 nt). The basic design (column circular CLUSTER) does not contain bystander solutions. The other guide RNAs contain G•U wobbles at positions −49, −27 and +48, as well as no bystander solution, G•A mismatch or depleted U at position −5. Data in a,b are shown as the mean editing percentage ± s.d. of n = 3 biological replicates.
Very recently, circular LEAPER guide RNAs were demonstrated to recruit endogenous ADARs with moderate editing efficiency and precision in cell culture and in vivo17,18. We benchmarked our best circular CLUSTER guide RNAs (Fig. 5a) for the Mecp2 W104>amber transcript against 111-nt-long symmetric LEAPER guide RNAs (Fig. 5b) and assessed various means of bystander editing suppression (Fig. 5 and Extended Data Fig. 5). Linear (Extended Data Fig. 5) and circularized LEAPER guide RNAs (Fig. 5b,c), using the Tornado expression system were tested by transfection into HeLa cells. As expected, the circular LEAPER-based design gave massive bystander editing at>10 sites, with yields of 20–39% at seven such sites (Fig. 5ccircular LEAPER). Again we first compared G•A mismatching16U depletion17 and G•U wobbles to suppress bystander editing at the four bystander sites, which are amenable to G•U wobble base pairing. Given the low number of amenable sites (Fig. 5c) the effects on on-target editing efficiency and bystander editing were comparably low. However, when we aimed to suppress off-target editing at the major ten bystander sites, the on-target yield of a pure G•A mismatch (17% ± 2%) or pure U depletion (16% ± 1%) solution dropped considerably, while a combination of G•U wobble and G•A mismatch (25% ± 2%) outperformed the combination of G•U wobble and U depletion (18% ± 1%) for the best-performing circular LEAPER guide RNA (Fig. 5c). In contrast, a linear CLUSTER guide RNA applying the G•U wobble strategy already achieved bystander-free 38% ± 7% on-target editing (Extended Data Fig. 5f). Furthermore, all optimized circular CLUSTER guide RNAs gave an exceptionally good editing efficiency of>84% on target with very good precision (Fig. 5d), clearly outcompeting all tested LEAPER guide RNAs (Extended Data Fig. 5). G•U wobbles entirely suppressed bystander editing at all three bystander sites where RSs bound the Mecp2 mRNA. It might be possible that G•A mismatches or U depletion would work similarly well to suppress bystander editing at such sites but this was not tested. There was one remaining site (5′-AAG) at position −5 with a moderate editing yield of 8% that was not amenable for wobble base pairing. Editing at this site would not change the amino acid sequence of MeCP2 yet its editing was reduced by U depletion or G•A mismatching (Fig. 5d).
In vivo proof-of-concept in a murine Rett syndrome model
Before applying the optimized circular CLUSTER guide RNA in vivo, we verified its successful circularization in cell culture with two sets of reverse transcription (RT)–qPCR primer pairs (Supplementary Fig. 11). The use of an outward primer pair (Extended Data Fig. 6a) allowed us to verify circularization with high confidence (Extended Data Fig. 6b,c), while the use of an inward primer pair (Extended Data Fig. 6d) allowed us to quantify the strong effect (235-fold increase) of circularization on the total guide RNA abundance (Extended Data Fig. 6e), suggesting that the majority of guide RNAs are fully processed (Extended Data Fig. 6f). We chose the PHP.eB serotype for adeno-associated virus (AAV) encapsulation as it allows cargo delivery to the mouse brain after systemic administration45. Indeed, this serotype was successfully used by us before to deliver the boxB/λ-ADAR tool into the same Rett syndrome mouse model for mutation correction44. The targeting virus encoded the circular CLUSTER guide RNA as displayed in Fig. 5a,d (G•U at G•U-amenable sites). For the nontargeting virus control, the guide RNA’s antisense parts were scrambled.
Mice were treated with 4 × 1012 viral genomes by retro-orbital injection and killed 4 weeks later; brain regions were analyzed separately for editing efficiency by Sanger sequencing. Editing levels differed among the seven brain regions, with clearly detectable editing in the midbrain, brainstem and thalamus, with an editing efficiency up to 19% (Fig. 6a). To better understand the key factors for successful editing, we analyzed all seven brain regions for the expression of the guide RNA (Fig. 6c), the AAV episome abundance (Fig. 6e) and the expression levels of all catalytically active murine Adar isoforms: total Adar1 (Fig. 6g), Adar1 p150 (Fig. 6i) and Adar2 (Fig. 6k). The relative expression of guide RNA and Adars were directly compared through RT–qPCR by normalization to the geometric mean of the same three housekeeping genes Actb, Rps29 and Rnu6 (Supplementary Figs. 12 and 13), while the AAV episome abundance was determined as the number of copies per cell. Unexpectedly, editing yield correlated the least with the Adar levels (Fig. 6h,j,l; R2 = 0.29–0.51), even though Adar expression differed among brain regions, particularly for Adar2 (for example, thalamus versus other brain regions). This indicates that Adar abundance did not limit the editing outcome. The strongest correlation was found between guide RNA expression level and editing yield (Fig. 6d; R2 = 0.87), suggesting that, even under circularization, guide RNA levels still limit on-target editing. Editing also correlated well with AAV abundance (Fig. 6f; R2 = 0.84) and, in most brain regions, guide RNA expression seemed to also correlate well with AAV abundance. However, there were a few exceptions, such as in the cortex, where guide RNA expression was low even though AAV abundance was comparably high. This indicates, in agreement with our previous findings44that the strength of the guide RNA promoter (for example, U6 promoter) or guide RNA stability differs among brain regions, thereby limiting editing even after successful viral delivery. In summary, these data show that the guide RNA’s abundance (determined by its delivery, expression and stability) is the most important factor to achieve high on-target editing in the brain, while Adar levels seem less important. This may instruct future designs of expression cassettes for CNS applications.

aEditing yields in different brain regions, after delivery of the AAV-PHP.eB-encoded guide RNA through retro-orbital injection into Rett mice carrying the Mecp2 W104>amber mutation and quantification by Sanger sequencing 4 weeks later. bCorrelation between the median editing in a and the geometric mean of the RT–qPCR targets in c,g,i,k. Olfactory bulb, Ob; cerebellum, Cb; hippocampus, Hi; cortex, Cx; midbrain, Mb; thalamus, Th; brainstem, Bs. cGuide RNA expression quantified by RT–qPCR (normalized to the geometric mean of Actb, Rps29 and Rnu6. dAs in b but correlating a,c. eAbsolute quantification of AAV episomes per cell by standard curve qPCR (normalized to Actb). fAs in b but correlating a,e. gAs in c but for Adar1. hAs in b but correlating a,g. iAs in c but for Adar1 p150. jAs in b but correlating a,i. kAs in c but for Adar2. lAs in b but correlating a,k. mBystander off-target events at the guide RNA-binding sites and the TS. nAll amplicon reads with on-target editing binned according to their number of bystander events (based on Supplementary Fig. 14). oGlobal RNA editing at 2,533 endogenous sites (coverage ≥ 50 reads, REDIportal55). pThalamus sections stained for MeCP2 and DAPI (nuclei). For adata are shown as the median editing percentage ± 95% CI determined in n = 3 mice (nontargeting virus) and n = 5 mice (targeting virus). For statistical analysis, a Mann–Whitney U-test (two-tailed, nonparametric) was applied. For c,e,g,i,k,m,ndata are shown as the median fold change ± 95% CI or median number of copies per cell ± 95% CI determined in n = 2 mice (three technical replicates each). The NGS analysis in o is based on results from n = 1 (nontargeting virus) or n = 2 (targeting virus) mice. The results in p are derived from n = 1 mouse per group. For b,d,f,h,j,lthe R² values were determined by simple linear regression.
Particularly in a clinical setting, editing must be efficient and precise, represented by cleanly edited transcripts devoid of unintended recoding events. To evaluate bystander off-target events in the Rett mouse model, we performed deep amplicon sequencing (average read depth of 47,009 and average coverage of 45,991) of the Mecp2 target transcript in all seven brain tissues. To ensure extensive detection of bystander events, we selected the two mice from the targeting virus group that had given the highest on-target editing yield in the thalamus. The on-target editing results matched very well with the Sanger sequencing (Fig. 6a,m and Extended Data Fig. 7). As controls, two nontargeting virus-treated mice were used. They showed considerably lower background (~0.2%) compared to Sanger sequencing (~5%) (Fig. 6a,m and Extended Data Fig. 7). Consequently, this now enabled us to measure on-target editing yields of 1.7–3.0% with high confidence in the olfactory bulb, cerebellum, hippocampus and cortex (Fig. 6m). Notably, bystander editing was hardly detected in any brain region. At three positions (−49, −27 and +48), G•U wobble base pairs were applied to suppress bystander editing. At positions −49 and −27, this was very successful; no bystander editing was detectable. At position +48, there may have been up to 0.12% bystander editing in the brainstem but 100-fold below the on-target editing yield in that tissue. Only one bystander was detected with high confidence. This was the unresolved, silent bystander site at position −5 that was already discovered in cell culture (Fig. 5d; 8% ± 4%). Here we detected up to 0.37% bystander editing in vivo in the brainstem (Fig. 6m). If they appear at many sites, even bystander events with low editing yield may sum up to interfere with on-target editing. To address this potential issue, we studied how often an on-target edited read is damaged by an additional bystander edit. We found that 98.3% of the ~10,000 detected on-target edited reads were completely bystander free (Fig. 6n and Supplementary Fig. 14), highlighting the impressive degree of editing precision achieved in the in vivo proof of concept.
Next, we evaluated the global editing precision by interrogating transcriptome-wide RNA sequencing (RNA-seq) data collected from the thalamus of targeting and nontargeting virus-treated Rett mice (Fig. 6o and Extended Data Fig. 8a). As before, we selected the two mice from the targeting virus group that gave the highest on-target editing yield in the thalamus with Sanger sequencing. First, we applied the RNA-editing index method that monitors changes in global RNA-editing levels in a highly unbiased manner and focused the analysis on the particularly critical coding sequence space46. The A-to-G RNA-editing indices were nearly identical in both groups (Extended Data Fig. 8a), indicating that the global editing activity was overall not affected by the presence of the targeting guide RNA. Second, we tried to detect differentially edited sites between the two conditions (Fig. 6o), similar to a previous study15. However, compared to the same analysis performed with untreated Rett mice (Extended Data Fig. 8b), we detected no clear off-target sites. Only three sites fell slightly outside of the ±25% Δ-editing margin but this was likely because of normal variability among mice. None of the three sites were located in the coding region and none of them were complementary to the guide RNA. Third, we tried to detect off-target events in a candidate approach and searched in silico throughout the whole murine genome for transcripts with 20-nt similarity (with up to one mismatch) to the guide RNA-binding regions. We identified 65 potential sites; however, only four of these sites were sufficiently expressed (≥20 reads of coverage) to be evaluated and no off-target editing was detected at any of them. Overall, we were unable to identify any global off-target events, excluding mouse-to-mouse variability, which suggests a very high precision of our approach on the transcriptome-wide level.
RNA base-editing approaches that apply the overexpression of an engineered editase, such as λN-ADAR2, typically suffer from substantial, editase-dependent global off-target events46,47,48. In our recent study—from which the noninjected mice data were derived (Extended Data Fig. 8a), we also detected numerous off-target events even though the native ADAR2 deaminase domain was fused to the λN peptide. We revisited this published RNA-seq data44 to compare with the CLUSTER generated data. The boxB/λN-ADAR2 data were generated using the same experimental setup (mouse model, AAV serotype and application route) as in this CLUSTER study. We ran the data in parallel through the same next-generation sequencing (NGS) analysis pipelines. First, we evaluated the same 2,533 endogenous RNA-editing sites (with coverage ≥ 50 reads) that we evaluated for the CLUSTER approach, as shown in Fig. 6o. Again, we selected the tissue with the highest on-target editing yield, which was the brainstem in this study44. We found a notable number (25) of potential off-target sites exhibiting a Δ-editing margin above 25% (Extended Data Fig. 8c). Importantly, the identified events included two evolutionary highly conserved, Adar2-specific editing sites in glutamate metabotropic receptor 4 (GRM4) and neuro-oncological ventral antigen 1 (NOVA1)49for which the change in editing level could have functional impact. Second, we analyzed the editing index in the coding sequence space and found a considerable increase in the index in the presence of the λN-ADAR2 deaminase (Extended Data Fig. 8c). Together, this shows, in accordance with the literature47that the harnessing of endogenous ADAR is more precise on the transcriptome-wide level than the ectopic expression of engineered ADAR effectors, whether hyperactive or native.
Lastly, we studied the restoration of MeCP2 protein expression and function upon treatment using the circular CLUSTER guide RNA (targeting virus). In the noninjected Rett mice, MeCP2 expression was not detectable by single-cell immunohistochemistry44 in the thalamus, as the mutation resulted in an unstable protein (Fig. 6p and Extended Data Fig. 9). In clear contrast, MeCP2 was restored in ~33.3% ± 4% (median ± 95% confidence interval (CI)) of the cells in the thalamus of the treated brain. Importantly, the restored MeCP2 protein was localized in heterochromatic foci inside the nucleus, which is an accepted proxy for its in vivo binding ability to methylated DNA50,51 and very similar to the positive control mouse where up to 100% of the cells showed MeCP2 protein associated with heterochromatin (Fig. 6p and Extended Data Fig. 9a,b). The abundance of foci increased with increasing editing yields over the evaluated brain tissues (Extended Data Fig. 9c). The results suggest that MeCP2 protein and its function are restored in cells where guide RNA is delivered and expressed.
Conclusions and outlook
The strategic placement of G•U wobble base pairs is a novel approach to improve editing precision and is widely applicable to RNA base-editing tools, including the LEAPER and CLUSTER approaches, λN-ADAR, Cas13-ADAR and chemically modified ASOs. The 5′-G•U wobble is particularly powerful to suppress editing at all four 5′-UAN triplets, which account for the largest burden of bystander editing. The suppressive effect of the G•U wobble is very strong and, at the same time, restricted to the direct 5′ and 3′ neighboring bases. In this regard, the G•U wobble differs from alternative solutions such as G•A mismatch or U depletion and makes the wobble strategy particularly powerful to suppress bystander editing close to an on-target site. As not all bystander-prone triplets are amenable to the G•U wobble strategy, we suggest to combine them with other strategies of bystander suppression, particularly with G•A mismatches at sites not amenable for the wobble strategy. Typically, but not always, a combination of wobble base pairing with G•A mismatching resulted in a better balance of editing efficiency over editing precision than the use of G•A mismatching only.
It is intriguing to speculate about the mechanism that underlies the suppressive effect of wobble base pairs. The ADAR deaminase domain strongly favors 5′-UAN triplets over 5′-CAN and 5′-GAN. A recent crystal structure analysis of the ADAR2 deaminase in complex with a dsRNA substrate shed light on the molecular basis of the nearest neighbor preference52 and identified a specific steric clash between the backbone of G489 and the exocyclic amino group of the guanine of the 5′ neighboring G≡C base pair (Extended Data Fig. 10a–c). Thus, replacing a 5′-A=U base pair with a 5′-G•U wobble base pair would again introduce a sterically demanding exocyclic amino group in the minor groove of the dsRNA substrate. Furthermore, the guanine base is likely further shifted toward the minor groove given the noncanonical hydrogen bond pattern in a G•U wobble base pair32. This might explain the very strong and located suppressive effect of the 5′ G•U wobble base pair (Extended Data Fig. 10a–c). A similar clash of the exocyclic amino group with S486 in the minor groove might explain the suppressive effect of the G•U wobble base pair at the 3′ nearest neighbor position (Extended Data Fig. 10d–f).
Overall, wobble base pairs create structural perturbations within the RNA substrate that have consequences for the activity of human ADAR. Moreover, regarding natural substrates, wobble base pairs seem to be part of a structural layer of control that may not yet be fully recognized. Nevertheless, we could deduce simple rules for the rational design of trans-acting guide RNAs for targeted RNA base editing, which achieved very high editing efficiency with very good editing precision. While broadly applicable in many approaches, the G•U wobble strategy proved helpful to design CLUSTER guide RNAs by means of expanding the sequence space for the computation of in silico optimized circular CLUSTER guide RNAs. In a direct comparison with LEAPER guide RNAs of similar length, we could show that CLUSTER guide RNAs can be designed with clearly advantageous efficiency and precision. It remains open whether G•A mismatching or U depletion could serve similarly well to optimize CLUSTER guide RNAs. However, given the short length of an individual RS (15–20 nt), we speculate that wobble base pairs might work better in highly A-rich binding regions where multiple G•A mismatches or bulged A bases might interfere with RS binding.
The AAV-mediated delivery of the most advanced circular CLUSTER guide RNA demonstrated effective (up to 19%) transcript repair of the Mecp2 W104>amber mutation in a mouse model of Rett syndrome and represents a successful in vivo recruitment of endogenous Adars for site-directed RNA editing in the CNS. With a median editing yield of ~14.4%, it also represents a high in vivo editing yield for restoring a disease-causing mutation in a murine model of human disease using only endogenous Adars. Furthermore, deep amplicon sequencing showed that on-target editing was achieved with very high precision regarding bystander editing. More than 98% of the detected on-target-edited reads were free from bystander edits. Only one (silent) bystander edit was detected with high confidence, albeit with a yield of only 0.1–0.37%. Transcriptome-wide RNA-seq confirmed a stable A-to-G RNA-editing index and no off-target sites in the transcriptome could be identified. This shows that editing can be achieved with very high precision and under the control of bystander editing in vivo. Notably, it is highly promising to see that endogenous Adar can be harnessed to obtain similar editing levels to those achieved with the exogenous boxB/λN-ADAR tool in the same mouse model before44. Interestingly, the trends regarding the varying editing efficiencies in different brain regions were also comparable between endogenous and exogenous ADAR approaches. A side-by-side analysis of on-target editing, AAV episome abundance as well as guide RNA and Adar isoform expression data53 suggests that not endogenous Ader expression but rather the virus-mediated guide RNA expression limited editing efficiency in the CNS of the murine Rett model. Thus, future improvements of AAV capsid engineering, application routes, guide RNA expression cassettes and/or guide RNA stability promise to enhance RNA base editing even further, breaking ground for clinical applications. Overall, harnessing endogenous ADAR with permanent AAV-driven CLUSTER guide RNAs in the CNS is an important next step toward the development of novel drug modalities that fight neurological diseases.
Methods
The GuideRNA-Forge tool
The GuideRNA-Forge tool was programmed in Python (version 3.9). It is based on our previously published ‘recruitment cluster finder’ tool and, thus, follows the core principles summarized previously15. GuideRNA-Forge uses a JavaScript Object Notation file (.json) to allow for the modular input of guide RNA parts and, thus, makes the power of in silico optimization available to any conceivable multivalent design, including circular guide RNAs. An example .json file including all relevant input values or strings for the generation of a circular CLUSTER guide RNA is provided in the GitHub archive.
The filters that identify binding regions for RS placement, while excluding RSs that contain unwanted sequences (for example, immunogenic, cytotoxic or guide RNA-destabilizing motifs) are now customizable. A combination of regular expressions (regex) and modular filter building blocks allows for quick assembly of user defined filters. The filters used in this study to generate G•U wobble-containing circular CLUSTER guide RNAs are provided in the GitHub archive. Note that the filters do not apply G•U wobbles to your guide RNA. Instead, they allow binding regions that contain A bases in a 5′-GAB, 5′-UAB, 5′-BAU (B = C, G or U), 5′-KAAU (K = G or U) and 5′-CAC sequence context, as well as A bases at the edges of binding regions. As not all A bases that allow for the G•U wobble solution (5′-UAB, 5′-BAU and 5′-KAAU) are necessarily edited, we recommend determining actual bystander off-target sites experimentally. G•U wobbles can then be strategically placed adjacent to problematic A bases.
In contrast to the previous version, the GuideRNA-Forge tool can identify multiple RSs within one potential binding region, as well as search in both the 5′ and the 3′ directions starting from the SD-binding site. Furthermore, the processing speed of recombination and folding (ViennaRNA package54) was drastically increased by implementing multithreading.
The resulting guide RNAs are scored for proximity of their binding sites andminimal secondary structure. CLUSTER guide RNAs with a high median score consist of RSs that bind in close proximity to each other on the target transcript and are less likely to engage in unproductive folding of their antisense part or misfolding of the ADAR-recruiting domain.
Vector design (guide RNA and cDNA)
The guide RNA and cis-acting reporter inserts including the necessary overhangs were created by hybridization and phosphorylation of oligonucleotides. The cis-acting editing reporters were created by cloning hybrid oligonucleotide inserts into our pcDNA3.1-based eGFP expression plasmid (pTS58) using ApaI and AgeI as described in Supplementary Note 1. Linear CLUSTER (using HindIII and BbsI into pTS1033), LEAPER (using HindIII and BamHI into pTS1033) and boxB (using HindIII and BamHI into pTS1033) guide RNAs were created as previously described15. DR guide RNAs were created by cloning hybrid oligonucleotide inserts encoding the antisense part into the PspCas13b crRNA backbone (Addgene plasmid 103854) using BbsI. The DR hairpin is already present in the backbone and must not be a part of the insert.
As the first step to create circular CLUSTER or LEAPER guide RNAs expressed by the U6 promoter, a modified Tornado expression cassette was created by gene synthesis (Thermo Fisher, GeneArt gene synthesis) and cloned into the backbone of our in-house editing vector ‘pEdit1.2’ (ref. 37) using NheI and AgeI, thereby replacing the ADAR2 expression cassette. The resulting Tornado OHA vector U6 (pTS1541) was used as the backbone for subsequent circular guide RNA cloning. The hybrid oligonucleotide guide RNA inserts were cloned into the latter backbone using BbsI. The cloning strategy is described in Supplementary Note 2. As a first step for cloning of circular LEAPER guide RNAs with flexible poly(AC) RNA linkers (AC50), the latter were synthesized as a gene block and cloned into pTS1541 using BbsI, resulting in the CircLEAPER AC50 cloning vector (pTS2508). Subsequent cloning of gene blocks containing circular LEAPER guide RNAs destined for the AC50 context was performed using BbsI into pTS2508. For the expression of circular guide RNAs by the EF1α promoter, the Tornado OHA vector EF1α (pTS1593) was used for subsequent circular guide RNA cloning analogous to pTS1541. For direct benchmarks between EF1α and U6 constructs, the hybrid oligonucleotide guide RNA inserts were cloned into pTS1593 (EF1α) and pTS1790 (U6) using BbsI. For stable genomic integration of linear or circular guide RNAs by the PiggyBac system, a transposase expression plasmid and the plasmid pTS1896 (transposon, U6, circular LEAPER guide RNA RAB7A), pTS1897 (transposon, U6, linear LEAPER guide RNA RAB7A), pTS1899 (transposon, EF1α, circular LEAPER guide RNA RAB7A) or pTS1900 (Transposon, EF1α, linear LEAPER guide RNA RAB7A) were used.
For AAV production, the lead circular CLUSTER guide RNA targeting the Mecp2 W104>amber transcript was cloned into the pAAV-GFP backbone (Cell Biolabs). The cloning strategy is described in Supplementary Note 3.
The open reading frame (ORF) sequence of murine Mecp2 W104>amber was supplied by the Mandel Lab (Vollum Institute, Oregon Health and Science University) and cloned into the pEGFP-N3 backbone (Clonetech) using EcoRI and KpnI under the control of a cytomegalovirus (CMV) promoter and terminated by an SV40 poly(A) signal. The human PEX1G843D plasmid was supplied by the Dodt Lab (Interfaculty Institute of Biochemistry and Cell Biochemistry, University of Tübingen). The cassette is under the control of an EF1α promoter and terminated by an EF1α poly(A) signal. The dual-luciferase reporters and the disease-relevant cDNA constructs AHI1 W725>amber, BMPR2 W298>amber and COL3A1 W1278>amber were created as previously described15. The sequences of all cloned products were verified by Sanger sequencing. The ORFs and amino acid sequences of Mecp2, PEX1 and the engineered editases are given in Supplementary Note 4. Plasmid maps of all mentioned pTS plasmids are given in Supplementary Note 5 or can be found in the literature37. Further details can be found in the Supplementary Information.
Analysis of RNA editing
A-to-I editing yields were quantified from Sanger sequence traces using SNAP-Gene (version 4.2.11). The relative height of the signal of G was compared to the sum of G+A, as described earlier37. If a reverse primer was used for sequencing, C and T peaks were treated accordingly. For better comparability, on-target editing yields of the same target with different guide RNAs were quantified using the same sequencing primer. Off-target editing had to be evaluated with different sequencing primers in most cases because of large distances between the guide RNA-binding sites. Only the cleanest reads were used for off-target evaluations, whereby G peaks below background were counted as 0% off target. Editing events with yields below 10% were background-corrected with the negative control.
Structural analysis
A-U base pair structures and ADAR amino acid residue structures were obtained from the substrate-bound ADAR2 dimer crystal structure (Protein Data Bank (PDB) 5HP2)56while G•U and U•G wobble base pairs were obtained from the NMR structure of the Gria2 stem loop RNA (PDB 2L2J)57. Structures were fitted and imaged with the open-source PyMOL Molecular Graphics System (version 2.5.0)58. The results are reported in Extended Data Fig. 10.
Cell culture
General
The used cell cultures were grown in DMEM (Thermo Fisher, 41965062) supplemented with 10% FBS (Thermo Fisher, 10270106) and kept in an incubator at 37 °C and 5% CO2.
Cell line choice
In our laboratory, HeLa cells have undergone comprehensive characterization, including assessment of ADAR1 expression, knockdown efficiency, response to interferon treatment and optimization of transfection conditions. Consequently, HeLa cells were used for most RNA base-editing experiments, particularly those involving editing of overexpressed exogenous transcripts. However, when targeting endogenous transcripts, achieving a high percentage of positively transfected cells is crucial for robust results. Each untransfected cell harbors the unedited transcript, which can adversely impact the overall readout. HEK293FT cells offer superior transfection efficiency and were, thus, used for targeting endogenous transcripts. By integrating a single genomic copy of ADAR1 p110, p150 or ADAR2 under the control of an CMV Tet-On promoter into Flp-In T-REx cells, we generated a set of cell lines that can be used to characterize how well dsRNA substrates such as bound guide RNAs or editing reporters are accepted by specific ADAR isoforms. Notably, the ADAR expression levels in these Flp-In cell lines exceeded typical endogenous levels, which increased bystander editing and facilitated the discovery of suppressive effects of wobble base pairs on bystander off-target edits.
ADAR Flp-In T-REx cells
A total of 2.5 × 105 ADAR1 p110 or p150 Flp-In T-REx cells or 3 × 105 ADAR2 Flp-In T-REx cells were seeded on poly(dLys)-coated 24-well plates in 500 µl of DMEM + 10% FBS + 10 ng ml−1 doxycycline. After 24 h, cells were transfected with 1,300 ng of cis-acting editing reporter plasmid (NucleoSpin Plasmid Transfection-grade, Macherey Nagel, 740490) using a 1:3 ratio of Lipofectamine 2000 (Thermo Fisher, 11668019). Then, 72 h after transfection, the cells were harvested. As the readout method, Sanger sequencing was used. Results are reported in Fig. 1Extended Data Figs. 1 and 3 and Supplementary Figs. 1 and 4.
HEK293FT cells
HEK293FT cells (6 × 104) were seeded in 24-well plates in 450 µl of DMEM + 10% FBS. After 24 h, cells were transfected with 1,200 ng of guide RNA plasmid (NucleoSpin Plasmid Transfection-grade, Macherey Nagel, 740490) using a 1:3 ratio of FuGene 6 (Promega, E2691). Then, 48 h after transfection, cells were harvested. As the readout method, Sanger sequencing was used. Results are reported in Extended Data Fig. 2 and Supplementary Figs. 2, 6 and 7.
HeLa cells (experiments using encodable guide RNAs)
A total of 0.8 × 105 HeLa cells were seeded in 24-well plates in 500 µl of DMEM + 10% FBS. Then, 24 h after seeding, cells were transfected with a total of 1,000 ng (800 ng of LEAPER or CLUSTER guide RNA plasmid and 200 ng of target-encoding plasmid) or a total of 1,200 ng (800 ng of boxB or DR guide RNA plasmid, 200 ng of target-encoding plasmid and 200 ng of λN-ADAR or Cas13-ADAR editase encoding plasmid) per well using a 1:1.5 ratio of plasmid to Lipofectamine 3000 ratio. Then, 72 h after transfection, cells were harvested. As the readout method, Sanger sequencing was used. Results are reported in Figs. 2–5Extended Data Figs. 3, 5 and 6 and Supplementary Figs. 5 and 11.
HeLa cells (experiments using an ASO guide RNA targeting the PEX1 G843D transcript)
A total of 1 × 105 HeLa cells were seeded in 24-well plates in 500 µl of DMEM + 10% FBS. Then, 24 h after seeding, cells were transfected with 300 ng of target PEX1G843D-encoding plasmid per well using a 1:3 ratio of plasmid to FuGene 6. Then, 48 h after seeding, the chemically modified guide RNAs were forward-transfected with 25 pmol of guide RNA and 1.5 µl of Lipofectamine RNAiMAX reagent (Thermo Fisher, 13778150) per well. Then, 24 h after guide RNA transfection, cells were harvested. The one-step RT–PCR (PEX1 primer set A) was followed by a nested PCR (PEX1 primer set B) for this specific target. As the readout method, Sanger sequencing was used. Results are reported in Fig. 3f.
HeLa cells (stable integration of guide RNAs)
A total of 1.5 × 105 HeLa cells were seeded in 12-well plates in 1 ml of DMEM + 10% FBS. Then, 24 h after seeding, cells were transfected with a total of 2,000 ng (1,600 ng of PiggyBac transposon vector containing the guide RNA expression cassette and 400 ng of pTS687 PiggyBac transposase vector) using a 1:3 ratio of plasmid to FuGene 6 (Promega, E2691). Then, 24 h after transfection, cells were transferred to 10-cm dishes and, after another 48 h, selection was started by addition of 5 µg ml−1 puromycin. Puromycin was refreshed on day 6 after transfection and selection was stopped 8 days after transfection. Each time when the cells were split, an aliquot was taken aside and used for RNA extraction and editing analysis. Results are reported in Supplementary Fig. 8.
HeLa cells (experiments using the dual-luciferase reporter system)
A total of 2.4 × 104 HeLa cells were seeded in 96-well plates. Cells were transfected 24 h after seeding with 160 ng of guide RNA plasmid and 40 ng of dual-luciferase reporter per well using a 1:1.5 ratio of plasmid to Lipofectamine 3000 and a 1:2 ratio of plasmid to P3000 reagent. For equimolar comparisons among differently sized guide RNA plasmids, their amount was adjusted accordingly and filled to a total of 160 ng with an empty pcDNA3.1 plasmid. The luciferase assay was performed 48 h after transfection. Results are reported in Supplementary Figs. 9 and 10.
Editing readout using dual-luciferase activity
Dual-luciferase activity was measured with the Dual-Luciferase Reporter Assay System (Promega) according to the manual in 96-well plates. Cells were washed with PBS and then lysed in 1× passive lysis buffer (35 µl per well) while shaking for 15 min at 700 r.p.m. at room temperature. Cell lysate (30 µl per well) was transferred to a LumiNunc 96-well plate (VWR, 732-2696) and measured in a Spark 10M plate reader (Tecan) using the dual-luciferase reporter assay reagents (35 µl per well) with an autoinjector. The attenuation standard settings (OD-none) of the Tecan reader were used. Each measurement was performed for 10 s, starting 5 s after injection. Per treatment, five biological replicates were analyzed, each measured in one technical replicate. For data processing, measured blank values (background) were subtracted from samples and controls and then all firefly values were divided by the corresponding On Ren values. The resulting normalized firefly activity of all samples was then set in ratio to the positive control to obtain the restored normalized firefly activity as a percentage.
Editing readout using Sanger sequencing (total RNA from cell lines)
Cells were harvested in RLT buffer (Qiagen, 79216), followed by RNA isolation using either the Monarch RNA cleanup kit (NEB, T2030L) or the RNeasy Mini RNA isolation kit (Qiagen, 74104). DNase I digestion was performed according to the manual using NEB DNase I (NEB, M0303S). One-step RT–PCR was performed using the Biotechrabbit one-step RT–PCR kit (Biotechrabbit, BR0400102) for regular substrates. For difficult substrates (for example, the murine Mecp2 W104>amber transcript), the OneTaq one-step RT–PCR kit (NEB, E5315S) was used. All samples were mixed with nuclease-free water (10 µl) and heated to 90 °C for 2 min immediately before RT–PCR. If necessary, a sense-oligo (1 µl, 10 µM) corresponding to the used guide RNA (Supplementary Table 2) was included in the volume. For the murine Mecp2 W104>amber transcript, the heating step was performed at 95 °C for 3 min under 12.5% DMSO (5% final concentration in RT–PCR mix). After TAE buffer–agarose gel electrophoresis and PCR cleanup (NucleoSpin Gel and PCR cleanup kit, Macherey Nagel, 740609), Sanger sequencing (Microsynth AG) was performed.
RT–qPCR experiments
RNA isolation was performed using either the Monarch RNA cleanup kit (NEB, T2030L) or the RNeasy Mini RNA isolation kit (Qiagen, 74104) according to the manufacturer’s protocol. DNase digestion was performed according to the manufacturer′s protocol (rigorous two-step incubation treatment) using the Turbo DNase Kit (Thermo Fisher, AM1907). RT with 500 ng of RNA per sample was performed using the high-capacity cDNA RT kit (Thermo Fisher, 4368814), followed by PCR cleanup using the NucleoSpin Gel and PCR cleanup kit (Macherey Nagel, 740609). RT–qPCR was executed in an Applied Biosystems QuantStudio 7 Pro real-time PCR system (96-well or 384-well qPCR plate, 20 ng or 4 ng of cDNA (10 or 2 ng μl–1) per well). The Fast SYBR-Green mastermix (Applied Biosystems, 4385612) was used according to the manufacturer’s protocol (10 µl or 5 μl of SYBR-Green mix, 7.2 µl or 2.6 μl of nuclease-free water and 0.4 μl or 0.2 µl of each primer). Samples and TE buffer negative controls were measured in three technical repl icates. ΔΔCt calculations were performed as previously described15. Amplification efficiency and melting curves were analyzed for each new primer pair using a cDNA dilution series (Supplementary Fig. 11). For normalization, one or more housekeeping genes from the following list were used: human U6 small nuclear RNA (snRNA; RNU6), murine U6 snRNA (Rnu6), murine β-actin (Actb) and murine ribosomal protein 29 (Rps29). Results are reported in Fig. 6 and Supplementary Figs. 11–13.
qPCR experiments (AAV episome copy number)
Genomic DNA isolation from murine brain tissue was performed using the QIAamp DNA Micro Kit (Qiagen, 56304) according to the manufacturer’s protocol. qPCR was executed in an Applied Biosystems QuantStudio 7 Pro real-time PCR system (384-well qPCR plate, 4 ng of genomic DNA (gDNA; 2 ng μl–1) per well). The Fast SYBR-Green mastermix (Applied Biosystems, 4385612) was used according to the manufacturer’s protocol (5 μl of SYBR-Green-Mix, 2.6 μl of nuclease-free water and 0.2 μl of each primer). Samples and AE buffer negative controls were measured in three technical replicates. ΔΔCt calculations were performed as previously described15. For normalization, the murine Actb housekeeping gene was used. The vector copy number per μg of DNA was determined by absolute quantification using a standard curve. The templates for the standard curve were prepared according to the manufacturer’s protocol (Takara, 6233). For the conversion of vector copy number per μg of DNA into copies per cell, 6.030 pg of gDNA was assumed per male C57BL/6 nucleus according to a previous study59. Results are reported in Fig. 6.
Animal experiments
Animal studies
All animal procedures were approved by the Institutional Animal Care and Use Committees of Oregon Health and Science University. A total of ten Mecp2G311A-carrying male mice (W104X) were killed during this study. Three mice were injected with the nontargeting virus and seven were injected with the targeting virus. From these seven animals, five were used for RT–qPCR and Sanger, amplicon and RNA-seq. The remaining two mice were used for MeCP2 immunohistochemistry.
Mecp2 311G>A husbandry and genotyping
All mice were housed in conventional laboratory housing under controlled humidity (target value, 44%; minimum, 24%; maximum, 64%), temperature (target value, 21 °C; minimum, 18 °C; maximum, 24 °C) and lighting (12-h light, 12-h dark period) with free access to food (regular mouse chow) and water. The Mecp2G311A/+ mice were maintained by crossing to pure wild-type C57BL/6J mice. Genotyping was performed using primers specific for the Mecp2G311A allele. Separately, sex was determined using PCR primers specific for the X and Y chromosomes (Supplementary Table 2).
Design of AAV-encoded guide RNAs
The targeting virus encoded the circular Mecp2-targeting CLUSTER guide RNA V27.2.4 (Supplementary Table 1). The latter consisted of a TS and four RSs of 20 nt length each, a split-R/G V3 ADAR-recruiting domain, a ligation stem and two ribozymes required for its circularization (Extended Data Fig. 4). For the nontargeting guide RNA virus, the sequences of all four RSs and the targeting sequence were scrambled, while A linkers, the ADAR recruiting motif and ribozymes required for circularization were unchanged (Supplementary Table 1).
Viral vector preparation
The AAV-PHP.eB vectors were produced by the Penn Vector Core Facility (Perelman School of Medicine, University of Pennsylvania) and titered using droplet digital PCR. Aliquots were stored at −80 °C before use.
Viral injections
P30–P34 male mice were deeply anesthetized with 3% isofluorane (v/v) and placed on a prewarmed surface. For each animal, a 100-μl volume containing 4E12 viral genomes was injected into the retro-orbital sinus. Following injections, mice were monitored for pain and distress while recovering on a heated pad before being returned to their home cage.
RNA isolation and Sanger sequencing (in vivo samples)
Four weeks after injection, brains were dissected in 5 mM HEPES in Hanks’ balanced salt solution and total RNA was isolated from individual brain regions using QIAzol reagent (Qiagen, 79306) and the Qiagen miRNeasy Kit reagent (Qiagen, 217004) according to the manufacturer’s instructions. RNA was reverse-transcribed using the SuperScript III first-strand synthesis system (Invitrogen, 18080051) and primed using oligo dT. Endogenous Mecp2 cDNA was amplified and analyzed by Sanger sequencing (Supplementary Table 2).
RT–qPCR experiments (in vivo samples)
Total RNA was isolated from individual brain regions using QIAzol reagent (Qiagen, 79306) and the Qiagen miRNeasy Kit (Qiagen, 217004) according to the manufacturer’s instructions. DNase digestion was performed on 1 μg of RNA using Turbo DNase Kit (Thermo Fisher, AM1907) according to the manufacturer’s instructions. DNase-treated RNA was then reverse-transcribed using the SuperScript III first-strand synthesis system (Invitrogen, 18080051) and was primed using oligo dT. RT–qPCR experiments were performed on an Applied Biosystems QuantStudio 6 Flex real-time PCR system. All samples were run in triplicate with a standard curve using SYBR select master mix reagent (Thermo Fisher, 4472918) for inward and outward primer sets to quantify CLUSTER guide concentrations across brain regions and circularization, respectively. CLUSTER guide expression was calculated relative to the geometric mean of three housekeeping genes (Rnu6, Rps29 and Actb). Primers sequences are listed in Supplementary Table 2.
Immunostaining
Immunostaining was performed as previously described60. Briefly, mice were anesthetized using an intraperitoneal injection of 2,2,2-tribromoethanol (Sigma Aldrich, T48402) and killed by transcardial perfusion of PBS pH 7.4, followed by 4% depolymerized paraformaldehyde. Brains were cryoprotected with sucrose, embedded in freezing medium and stored at −80 °C. Sagittal whole-brain sections were cut at 25 μm using a cryostat and stored at −20 °C until staining. Sections underwent heat-mediated antigen retrieval before blocking in PBST (0.1% Triton X-100 in PBS, pH 7.4) and 10% BSA (Sigma, A2153-500G) for 1 h at room temperature. Sections were incubated overnight at 4 °C with rabbit anti-MeCP2 antibody (rabbit monoclonal antibody D4F3, RRID: AB_2143849, Cell Signaling, 1:500) diluted in blocking buffer. Sections were washed with PBST and incubated for 1 h at room temperature with Alexa Fluor 488 donkey anti-rabbit IgG (Invitrogen, A21206 1:750) diluted in blocking buffer. Sections were washed and incubated with 300 nM DAPI (Invitrogen, D1306) in PBS for 5 min. After a final wash in PBS, sections were mounted using Fluoromount G (Invitrogen, 00-4958-02). Images were acquired using ZEN 2.3 SPI FPS Black (version 14.0.0.0) on a Zeiss LSM 710 confocal microscope with a C-Apochromat ×40 (1.2 numerical aperture) objective and LSM T-PMT detector. Images were processed in ImageJ (version 1.54f). Murine MeCP2 protein and nuclei (DAPI) were pseudo-colored in ImageJ to highlight colocation puncta. Results are reported in Fig. 6p and Extended Data Fig. 9.
Deep amplicon NGS experiment
Retro-orbital injections and RNA isolation were performed as explained above. Two settings were carried out, each with an independent duplicate: (1) nontargeting circular CLUSTER guide RNA and (2) Mecp2 W104>amber transcript-targeting circular CLUSTER guide RNA. For the latter we selected the two thalamus samples that showed the highest Mecp2 on-target editing yield during Sanger sequencing. Purified RNA was delivered to the Massively Parallel Sequencing Shared Resource Core (MPSSR) at the Oregon Health and Science University for Illumina library preparation and then transferred to the Molecular Technologies Core (MTC) at the Oregon National Primate Research Center for Illumina amplicon sequencing. The library was prepared with the TruSeq DNA nano library prep kit and sequenced with a MiSeq instrument (1 million reads, 500 cycles, 2 × 500 bp of paired-end reads; Illumina). Furthermore, 2 nM pooled libraries were spiked with 5% PhiX before the sequencing run. The sequencing reads were demultiplexed using BCL Convert (version 2.4.0). Adaptors were trimmed with FastQ (version 1.0.0). Raw FASTQ files were processed using Seqtk (version 1.3-r106). The command ‘seqtk trimfq in.fastq> out.fastq’ was used to trim low-quality bases. The command ‘seqtk seq -q30 -n N in.fastq> out.fastq’ was used to mask base calls with a quality value < 30 as N. The base-call accuracy of the remaining bases was, thus, 99.9%. FASTQ file quality was assessed with FastQC (version 0.11.9)61.
Mapping of deep amplicon-seq reads
Reads were aligned to the GRCm38/mm10 reference genome using BWA-MEM (version 0.7.17-r1188). The alignments were analyzed using the Integrative Genomics Viewer (version 2.16.2). The editing yields were exported from IGV (version 2.16.2) and then further processed in Excel 2016.
Next-generation RNA-seq experiment
Retro-orbital injections and RNA isolation were performed as explained above. Two settings were carried out, each with an independent duplicate: (1) nontargeting circular CLUSTER guide RNA and (2) Mecp2 W104>amber transcript-targeting circular CLUSTER guide RNA. For the latter, we selected the two thalamus samples that showed the highest Mecp2 transcript on-target editing yield during Sanger sequencing. Purified RNA was delivered to CeGaT for total RNA-seq. The library was prepared from 100 ng of RNA with the KAPA RNA HyperPrep library prep kit with RiboErase (HMR) and KAPA globin depletion hybridization oligos (Roche) and sequenced with a NovaSeq 6000 (50 million reads, 2 × 100 bp of paired-end reads; Illumina). The sequencing reads were demultiplexed using Illumina bcl2fastq (version 2.20). Adaptors were trimmed with Skewer (version 0.2.2)62. No quality trimming of the reads was performed. Raw FASTQ file quality was assessed with FastQC (version 0.11.8)61. One of the nontargeting CLUSTER guide RNA samples failed quality control (RNA integrity number of 1) and, thus, had to be removed from the analysis. Results are reported in Fig. 6o and Extended Data Fig. 8. In addition, we reanalyzed brainstem samples from a similar previous study that applied the boxB/λN-ADAR system44. From this existing dataset, we randomly selected samples from the untreated Rett mouse negative control group and the two samples from the boxB/λN-ADAR targeting virus-treated group that that showed the highest Mecp2 transcript on-target editing yield. Results are reported in Extended Data Fig. 8.
Mapping of RNA-seq reads
Reads were uniquely aligned to the GRCm38/mm10 reference genome using STAR (version 2.7.3a) (‘STAR-2.7.3a –alignSJoverhangMin 8 –alignIntronMax 1000000 –alignMatesGapMax 600000 –outFilterMismatchNoverLmax 0.3 –outFilterMismatchNoverReadLmax 1 –outFilterMatchNminOverLread 0.66 –outFilterMultimapNmax 1 –outReadsUnmapped Fastx –outSAMattributes All –outSAMtype BAM Unsorted –quantMode GeneCounts –genomeLoad LoadAndKeep –limitBAMsortRAM 37580963840 –outBAMsortingThreadN 10 –runThreadN 40 –genomeDir mm1 0.STAR.7.ReadsLn100.gencodeM18 –readFilesCommand cat –readFilesIn file-name_R1.fastq file-name_R2.fastq –outFileNamePrefix file-name’)63.
RNA-editing index
The RNA-editing index tool46 calculates the average editing level across all A bases in a set of regions. The editing index is defined as the ratio of the number of A•G mismatches to the total coverage of A bases. Genomic sites overlapping common single-nucleotide polymorphisms (dbSNP142) were excluded. The RNA-editing index tool was used to assess the overall editing for all the coding sequence regions of the mouse genome.
Global RNA editing
To assess global RNA editing, we used the known editing sites from the REDIPortal55 and from a very small set of evolutionarily conserved A-to-I editing sites64. We calculated the editing levels in those sites using REDITools65 with a coverage ≥ 50 reads for every site. For every group of each dataset (this study and the previous study44), we combined the editing levels of the replicates for each site. We then selected the 2,532 sites that were common in both datasets.
Data analysis
Data were analyzed using Excel 2016 and GraphPad Prism 8. Figures were created with CorelDraw 2017. The manuscript was written using Word 2016. The custom GuideRNA-Forge tool was written in Python (version 3.9). Guide RNA folds were created using the ViennaRNA Package (version 2.0, http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi). qPCR analysis was performed using the Applied Biosystems 7500 data analysis software version 1.1 (animal experiment) and version 2.3 (remainder of the study).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Transcriptome-wide RNA-seq data are accessible from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus database with accession code GSE265898. Deep amplicon NGS data are accessible from the NCBI Sequence Read Archive SRA database with accession code PRJNA1100948. Transcriptome-wide RNA-seq data from Sinnamon et al. are accessible from the NCBI SRA database with accession code PRJNA849938. Structural data from Thuy-Boun et al. and Stefl et al. are available from PDB 5HP2 and 2L2Jrespectively.
Code availability
The full code of the GuideRNA-Forge tool is available upon request. The compiled tool can be obtained from GitHub (https://github.com/recruitment-cluster-finder/GuideRNA-FORGE)66.
References
-
Diaz Quiroz, J. F., Siskel, L. D. & Rosenthal, J. J. C. Site-directed A → I RNA editing as a therapeutic tool: moving beyond genetic mutations. RNA 29498–505 (2023).
-
Rees, H. A. & Liu, D. R. Base editing: precision chemistry on the genome and transcriptome of living cells. Night. Rev. The gene. 19770–788 (2018).
-
Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 1783–96 (2016).
-
Bass, B. L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 71817–846 (2002).
-
Stafforst, T. & Schneider, M. F. An RNA–deaminase conjugate selectively repairs point mutations. Angew. Chem. Int. Ed. Engl. 5111166–11169 (2012).
-
Vogel, P. et al. Efficient and precise editing of endogenous transcripts with SNAP-tagged ADARs. Nat. Methods 15535–538 (2018).
-
Montiel-Gonzalez, M. F. et al. Correction of mutations within the cystic fibrosis transmembrane conductance regulator by site-directed RNA editing. Proc. Natl Acad. Sci. USA 11018285–18290 (2013).
-
Montiel-Gonzalez, M. F., Vallecillo-Viejo, I. C. & Rosenthal, J. J. An efficient system for selectively altering genetic information within mRNAs. Nucleic Acids Res. 44e157 (2016).
-
Vallecillo-Viejo, I. C. et al. Abundant off-target edits from site-directed RNA editing can be reduced by nuclear localization of the editing enzyme. RNA Biol. 15104–114 (2018).
-
Cox, D. B. T. et al. RNA editing with CRISPR–Cas13. Science 3581019–1027 (2017).
-
Abudayyeh, O. O. et al. A cytosine deaminase for programmable single-base RNA editing. Science 365382–386 (2019).
-
Kannan, S. et al. Compact RNA editors with small Cas13 proteins. Nat. Biotechnol. 40194–197 (2022).
-
Merkle, T. et al. Precise RNA editing by recruiting endogenous ADARs with antisense oligonucleotides. Nat. Biotechnol. 37133–138 (2019).
-
Monian, P. et al. Endogenous ADAR-mediated RNA editing in non-human primates using stereopure chemically modified oligonucleotides. Nat. Biotechnol. 401093–1102 (2022).
-
Reautschnig, P. et al. CLUSTER guide RNAs enable precise and efficient RNA editing with endogenous ADAR enzymes in vivo. Nat. Biotechnol. 40759–768 (2022).
-
Qu, L. et al. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 371059–1069 (2019).
-
Yi, Z. et al. Engineered circular ADAR-recruiting RNAs increase the efficiency and fidelity of RNA editing in vitro and in vivo. Nat. Biotechnol. 40946–955 (2022).
-
Katrekar, D. et al. Efficient in vitro and in vivo RNA editing via recruitment of endogenous ADARs using circular guide RNAs. Nat. Biotechnol. 40938–945 (2022).
-
Eggington, J. M., Greene, T. & Bass, B. L. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2319 (2011).
-
Licht, K. et al. Inosine induces context-dependent recoding and translational stalling. Nucleic Acids Res. 473–14 (2019).
-
Wong, S. K., Sato, S. & Lazinski, D. W. Substrate recognition by ADAR1 and ADAR2. RNA 7846–858 (2001).
-
Higuchi, M. et al. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron–exon structure determines position and efficiency. Cell 751361–1370 (1993).
-
Huang, H. et al. Tissue-selective restriction of RNA editing of CaV1.3 by splicing factor SRSF9. Nucleic Acids Res. 467323–7338 (2018).
-
Hanrahan, C. J. et al. RNA editing of the Drosophila for Already+ channel transcript. Evolutionary conservation and developmental regulation. Genetics 1551149–1160 (2000).
-
Reenan, R. A., Hanrahan, C. J. & Ganetzky, B. The mlenapts RNA helicase mutation in Drosophila results in a splicing catastrophe of the for Already+ channel transcript in a region of RNA editing. Neuron 25139–149 (2000).
-
Ingleby, L. et al. Regulated RNA editing and functional epistasis in Shaker potassium channels. J. Gen. Physiol. 13317–27 (2009).
-
Reenan, R. A. Molecular determinants and guided evolution of species-specific RNA editing. Nature 434409–413 (2005).
-
Ramaswami, G. et al. Genetic mapping uncovers cis-regulatory landscape of RNA editing. Nat. Commun. 68194 (2015).
-
Schneider, M. F. et al. Optimal guideRNAs for re-directing deaminase activity of hADAR1 and hADAR2 in trans. Nucleic Acids Res. 42e87 (2014).
-
Uzonyi, A. et al. Deciphering the principles of the RNA editing code via large-scale systematic probing. Mol. Cell 812374–2387 (2021).
-
Liu, X. et al. Learning cis-regulatory principles of ADAR-based RNA editing from CRISPR-mediated mutagenesis. Nat. Commun. 122165 (2021).
-
Varani, G. & McClain, W. H. The G•U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep. 118–23 (2000).
-
Xu, D. et al. The electrostatic characteristics of G•U wobble base pairs. Nucleic Acids Res. 353836–3847 (2007).
-
Stefl, R. et al. Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure 14345–355 (2006).
-
Chahrour, M. et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 3201224–1229 (2008).
-
Heep, M. et al. Applying human ADAR1p110 and ADAR1p150 for site-directed RNA editing—G/C substitution stabilizes guideRNAs against editing. Genes (Basel) 834 (2017).
-
Wettengel, J. et al. Harnessing human ADAR2 for RNA repair—recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 452797–2808 (2017).
-
Dixon-Salazar, T. et al. Mutations in the AHI1 gene, encoding Jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 75979–987 (2004).
-
Smith, L. T. et al. Mutations in the COL3A1 gene result in the Ehlers–Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. J. Invest. Dermatol. 108241–247 (1997).
-
Roberts, KE et al. BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur. Respir. J. 24371–374 (2004).
-
Davis, A. R. & Znosko, B. M. Thermodynamic characterization of single mismatches found in naturally occurring RNA. Biochemistry 4613425–13436 (2007).
-
Gartner, J. et al. Mutations in PEX1 in peroxisome biogenesis disorders: G843D and a mild clinical phenotype. J. Inherit. Metab. Dis. 22311–313 (1999).
-
Litke, J. L. & Jaffrey, S. R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 37667–675 (2019).
-
Sinnamon, J. R. et al. Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett syndrome. Proc. Natl Acad. Sci. USA 119e2206053119 (2022).
-
Chan, K. Y. et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 201172–1179 (2017).
-
Roth, S. H., Levanon, E. Y. & Eisenberg, E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat. Methods 161131–1138 (2019).
-
Pfeiffer, L. S. & Stafforst, T. Precision RNA base editing with engineered and endogenous effectors. Nat. Biotechnol. 411526–1542 (2023).
-
Buchumenski, I. et al. Global quantification exposes abundant low-level off-target activity by base editors. Genome Res. 312354–2361 (2021).
-
Pinto, Y., Cohen, H. Y. & Levanon, E. Y. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol. 15R5 (2014).
-
Heckman, L. D., Chahrour, M. H. & Zoghbi, H. Y. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife 3e02676 (2014).
-
Brown, K. et al. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum. Mol. Genet. 25558–570 (2016).
-
Matthews, M. M. et al. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat. Struct. Mol. Biol. 23426–433 (2016).
-
Lein, E. S. et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445168–176 (2007).
-
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 626 (2011).
-
Picardi, E. et al. REDIportal: a comprehensive database of A-to-I RNA editing events in humans. Nucleic Acids Res. 45D750–D757 (2017).
-
Thuy-Boun, A. S. et al. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. 487958–7972 (2020).
-
Stefl, R. et al. The solution structure of the ADAR2 dsRBM–RNA complex reveals a sequence-specific readout of the minor groove. Cell 143225–237 (2010).
-
Baugh, E. H. et al. Real-time PyMOL visualization for Rosetta and PyRosetta. PLoS ONE 6e21931 (2011).
-
Capparelli, R. et al. DNA content differences in laboratory mouse strains determined by flow cytometry. Cytometry 29261–266 (1997).
-
Sinnamon, J. R. et al. In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 32107878 (2020).
-
Andrews, S. FastQC: a quality control tool for high-throughput sequence data. Available online at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. (2010).
-
Jiang, H. et al. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinf. 15182 (2014).
-
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2915–21 (2013).
-
Gabay, O. et al. Landscape of adenosine-to-inosine RNA recoding across human tissues. Nat. Commun. 131184 (2022).
-
Lo Giudice, C. et al. Investigating RNA editing in deep transcriptome datasets with REDItools and REDIportal. Nat. Protoc. 151098–1131 (2020).
-
recruitment-cluster-finder / GuideRNA-FORGE. GitHub https://github.com/recruitment-cluster-finder/GuideRNA-FORGE (2024).
Acknowledgements
We gratefully acknowledge generous support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; STA 1053/10-1 and STA 1053/11-1 to T.S.) and the Baden-Württemberg Ministry of Science as part of the Excellence Strategy of the German Federal and State Governments with an intramural innovation grant (IG-Reauts-2019-05 to P.R.). Further generous support was provided by the International Rett Syndrome Foundation (grant 3806 to T.S.), by the VolkswagenStiftung (Momentum Grant 96 876 to T.S.) and by the National Institutes of Health (grant NS110868 to G.M.). E.Y.L. was supported by the Israel Science Foundation grants 2039/20 and 2637/23. We also want to thank J. J. C. Rosenthal and J. D. Quiroz (Eugene Bell Center for Regenerative Biology and Tissue Engineering, The Marine Biological Laboratory) and G. Dodt (Interfaculty Institute of Biochemistry, University of Tübingen) for supplying us with the plasmids encoding the λN-ADAR editases and PEX1G843D that were applied in Fig. 3. In addition, we want to thank J. B. Li and I. Jarmoskaite (Department of Genetics, Stanford University) for insights and discussion on engineering improved ADAR recruitment motifs. Lastly, we want to acknowledge technical support from the CeGaT GmbH (Illumina RNA-seq), from the MPSSR at the Oregon Health and Science University (Illumina library preparation) and from the MTC at the Oregon National Primate Research Center (Illumina amplicon sequencing).
Ethics declarations
Competing interests
T.S., G.M. N.W., C.P.W., L.S.P., A.E.S. and P.R. hold patents on site-directed RNA editing. T.S., C.P.W., A.E.S. and P.R. are inventors of a filed patent based on the work published here. T.S. is a cofounder and shareholder of AIRNA Bio. G.M. is a cofounder and shareholder of Vico Therapeutics. The other authors declare no conflicts of interest.
Peer review
Peer review information
Nature Biotechnology thanks Michael Jantsch and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 The enhancing and suppressing effects of G·U and U·G wobble base pairs on RNA editing apply to all catalytically active ADAR isoforms.
(a) Schematic of the utilized cis-acting editing reporter construct. (b) Editing yields when applying G·A mismatches or G·U wobbles for editing suppression at the 5’-UAG triplet in either ADAR1 p110-, ADAR p150- or ADAR2-Flp-In T-REx cells. The exact position of each mismatch or wobble is indicated below each bar. The target adenosine is highlighted in green, bold and underlined. Bases that induce wobbles or mismatches are highlighted in dark blue and bold. The 5’ and 3’ orientation of the triplet and the used ADAR isoform are shown above the grouped bars. (c) As b) but regarding editing yields when applying enhancing U·G wobbles. Data for b) and c) are shown as the mean ± s.d. of N = 3 biological replicates per bar. For statistical analysis, a student’s t-test (two-tailed, parametric) was applied.
Extended Data Fig. 2 G·U wobbles improve efficiency and precision of trans-acting LEAPER guide RNAs for endogenous targets.
(a-b) Editing heat-map of the LEAPER guide RNA binding site within the indicated transcripts. The basic design column, LEAPER, lacks G·A mismatches and wobble base pairs. The other guide RNAs contain either G·A mismatches or G·U wobbles at G·U amenable sites or a combination of both solutions at all bystander sites. In the latter case, G·A mismatches are placed at sites not amenable to G·U wobbles. The triplet context for each listed editing event is given with the target adenosine highlighted in green and all off-target adenosines in blue. The position of each site is given rela tive to the transcript and the target adenosine (±0 position). The editing events detected at position 2546 ( + 47) and 2548 ( + 49) of the NUP43 transcript were natural events and were thus skipped in regard to bystander suppression. Editing was performed with plasmid-borne guide RNA and endogenous target transcripts in HEK293FT cells (endogenous ADAR). Data are shown as the mean editing percentage ± s.d. of N = 3 biological replicates.
Extended Data Fig. 3 G·U wobbles significantly outperform previous solutions in bystander suppression at nearest neighbor (NN) sites.
(a) Design of the cis-acting RNA editing reporter constructs used in e) and f). (b) Design of the linear trans-acting CLUSTER guide RNAs used in g). (c) Illustration of the base-pairing motifs applied to the constructs in a) (ocher) and b) (grey) that were used to suppress bystander editing at the 5´-U[[AAG]sequence motif in e) and g). (d) As c) but for the 5´-[U[UAA]U sequence motif and the results in f). (e) Suppression of bystander editing at the 5´-NN A and concurrent effect on the target A editing yield in a 5’-U[[AAG]sequence motif. (f) Similar to e) but targeting the A in a 5’-[U[UAA]U sequence motif and suppressing bystander editing at a 3´-NN A. Editing in e) and f) was performed in ADAR1 p110 Flp-In T-REx cells using plasmid-borne reporters as displayed in a). (g) Suppression of bystander editing at a 5´-NN A when editing a 5’-U[[AAG]site (K984) of the human BMPR2 transcript using trans-acting CLUSTER guide RNAs. (h) Meta-analysis of bystander suppression at 5´- and 3´-NN sites over three different target transcripts (AHI1, BMPR2 and COL3A1), three different A-rich target triplets flanked either 5’ and/or 3’ by uridines (5’-[U[UAA]U, 5’-U[[AAG], 5’-U[[AAA]U), and applying two trans-acting site-directed RNA editing systems (CLUSTER/endogenous ADAR, boxB/λN-ADAR2Q). The full data set of the meta-analysis is shown in Supplementary Fig. S5. Editing in g) and h) was performed in HeLa cells using plasmid-borne editase (λN-ADAR2Q) and/or guide RNA (2x boxB, CLUSTER). Data in e) – g) are shown as the mean editing percentage ± s.d. of N = 3 biological replicates. Data in h) are shown as the mean editing percentage ± s.d. of three 5’-NN off-target sites, seven on-target sites, and five 3’-NN off-target sites containing N = 3 biological replicates per site. The center line represents the median, the plus-sign the mean. The whiskers extend to the minimum and maximum values. The box limits the 25th and 75th percentile. For statistical analysis, a student’s t-test (two-tailed, parametric) was applied.
Extended Data Fig. 4 Circularization of a split-R/G CLUSTER guide RNA using the Tornado expression system.
The guide RNA is expressed from either a polymerase III (for example U6) or a polymerase II (for example CAG) promoter with a corresponding terminator. 5’ and 3’ ribozymes auto-cleave the primary transcript resulting in a 5’-OH and a 2’,3’-cyclic phosphate overhang, respectively, which are subsequently ligated by the endogenous RTCB ligase. In the final guide RNA, the ligation stem (orange) forms a nearly continuous duplex with the split-R/G motif (light green), only interrupted by a five-nucleotide bulge. The circular guide RNA binds to its target transcript. Strategic placement of wobble base pairs within the recruitment sequences (RS) and/or the specificity domain (dark green) suppresses off-target editing, while a C-A mismatch within the specificity domain (SD) promotes on-target A-to-I RNA editing.
Extended Data Fig. 5 Extended data set for editing Mecp2 W104amber with LEAPER and CLUSTER guide RNAs.
Panels (a) to (d) give schematic representations of the used linear and circular LEAPER and CLUSTER guide RNAs and their binding to the target mRNA. LEAPER guide RNAs were 111 nt long, centered with a C-A mismatch on the target adenosine. CLUSTER guide RNAs combined an ADAR-recruiting domain (R/G motif), a 20 nt long specificity domain containing the C-A mismatch with the target adenosine at position 8, and a cluster of three recruitment sequences of 20 nt length each. The circular CLUSTER guide RNA combines the ligation stem for circularization with the ADAR-recruiting motif into a split-R/G motif. (e) Editing heat-map using linear and circular LEAPER guide RNAs as seen in a) and b). (f-h) Editing heat map using linear and circular CLUSTER guide RNAs as seen in c) and d). Notably, the linear R/G version 21 guide RNA achieved already highly precise editing with an impressive 38 ± 7% on-target editing yield, clearly better than the best circular LEAPER guide RNA design (25 ± 2% on-target yield) with similar precision. A circular CLUSTER guide RNA with six recruitment sequences, but lacking the ADAR recruitment motif, achieved 54 ± 7% on-target editing with very high precision after applying the GU wobble strategy. However, adding a split-R/G recruiting motif achieved even better on-target editing yields of 75 ± 1% to 82 ± 1%, even though only 4 recruitment sequences were applied. GU wobbling was required to eliminate bystander editing at the binding sites of recruitment sequences. The triplet context for each listed editing event is given with the edited adenosine highlighted in bold green. The position of each bystander site (bold blue) is given relative to the target adenosine (±0 position). Editing was performed with plasmid-borne guide RNA and murine Mecp2 in HeLa cells (endogenous ADAR). Data are shown as the mean editing percentage ± s.d. of N = 3 biological replicates.
Extended Data Fig. 6 Verifying guide RNA circularization via quantitative RT-qPCR.
The circular state of the split-R/G CLUSTER guide RNA (V27.2.4, pTS2108) targeting mMecp2 W104Amber was verified by using quantitative RT-PCR on in vitro samples from transfected HeLa cells. As reference a linear version of the same guide RNA was used. The dataset was normalized to human RNU6 snRNA. The amplicon of the outward primer pair in a) is 187 bp long and requires reverse transcription and PCR through a strong hairpin structure. It can thus not be directly compared to the inward primer pair in d) which is smaller (75 bp) and does not contain inhibitory secondary structures. (a) Binding sites of the outward primer pair, which can only amplify circular guide RNAs. (b) Fold change of linear and circular split-R/G CLUSTER guide RNAs (V27.2.4, pTS2108, pTS2228) relative to endogenous human RNU6 as detected by using the outward primer pair. The 2984-fold difference between linear and circular shows that the endogenous circularization of the guide RNAs though the RTCB ligase (Extended Data Fig. 4) was successful and that the outward primer pair can identify circular guide RNAs with high confidence (low type-I error rate). (c) Type-I error rate calculated from b) amounts to only 0.03%, thus 99.97% of circular guide RNAs are correctly identified. (d) Binding sites of the inward primer pair, which can amplify both linear and circular guide RNAs. (e) Fold change of linear and circular split-R/G CLUSTER guide RNAs (V27.2.4, pTS2108) relative to endogenous human RNU6 snRNA as detected by using the inward primer pair. The 235-fold difference between linear and circular shows the high level of total guide RNA enrichment achieved after circularization. (f) Percentage of successfully circularized guide RNAs calculated from e). Under the assumption that the 235-fold increase detected for circular guide RNAs compared to linear ones solely depends on circularization, 99.6% of all guide RNAs were successfully processed by the ribozymes of The tornado expression system and then ligated by RTCB. Data in b) and e) are shown as the mean editing percentage ± s.d. of N = 3 biological replicates.
Extended Data Fig. 7 Amplicon sequencing results before background correction.
(a) Editing heat map of the indicated brain tissue of Rett syndrome mice treated with either the scrambled circular CLUSTER guide RNA virus (nontargeting virus) or (b) the circular CLUSTER guide RNA virus (targeting virus). The triplet context for the target-site row is boxed, and the target adenosine underlined and green. All other triplet contexts contain an off-target adenosine in blue. The position of each site is given relative to the transcript and the target adenosine (±0 position). The dataset covers the complete guide RNA binding region including all binding sites (BS) and the target sequence (TS) containing the target adenosine. Data are shown as the mean editing percentage of N = 2 mice.
Extended Data Fig. 8 Analysis of global off-target events by RNA-seq.
(a) Rett mice injected with wobble-optimized circular CLUSTER guide RNA (targeting virus) versus scrambled guide RNA (nontargeting virus). (a) (top) A-to-G RNA editing index in coding sequences. An index of 1 means 1% of RNA nucleotides mapped to genomic adenosines are guanosines. The index was stable (no change in global editing activity due to the targeting guide RNA). (a) (bottom) Global RNA editing at 2,528 endogenous sites (coverage ≥ 50, REDIportal55). With only 3 sites showing > 25% Δ-editing (beyond blue dotted lines), no clear off-target editing was detectable in comparison to panel b. (b) Reanalyzed RNA-seq data (previous study44). With respect to the tissue selected in panel c), data from the brainstems of two untreated Rett mice are compared. (b) (top) Both untreated mice show a stable editing index. (b) (bottom) Global RNA editing at 2,301 endogenous sites (coverage ≥ 50). These sites are also displayed in panel a) (bottom) and c) (bottom). The 7 sites showing > 25% Δ-editing are considered normal mouse to mouse and technical variability. (c) Reanalyzed RNA-seq data (previous study44). Rett mice injected with boxB/λN-ADAR2 virus were compared to untreated ones. The ADAR2 catalytic domain in the virus is from the native ADAR2 protein. (c) (top) The A-to-G index increase by 0.007 in the coding space reflects a substantial and somewhat concerning increase in global editing, probably due to the overexpressed ADAR2 catalytic subunit. (c) (bottom) Global RNA editing at 2,533 endogenous sites that include the 2,528 sites shown in panel a) (coverage ≥ 50 reads). 25 sites showing > 25% Δ-editing after injection of the λN-ADAR2 targeting virus. This is in clear contrast to the very high precision seen for CLUSTER guide RNAs recruiting endogenous ADARs in a) (bottom) or the untreated mice in b) (bottom). Importantly, the potential off-target sites included two ADAR2-specific, highly evolutionary conserved sites in GRM4 (glutamate transmission) and NOVA1 (splicing)49. Data in a), b) and c) are based on results from N = 1-2 mice per group. Thalamus and brainstem were selected for showing the highest editing yields in their respective study.
Extended Data Fig. 9 Immunohistochemical evidence supporting repair of MeCP2 protein and function after treatment of Rett mice with the circular CLUSTER guide RNA.
(a) Confocal images of sections of thalamus stained for MeCP2 pr otein and DAPI to indicate nuclei. MeCP2 puncta staining, co-localizing with DAPI, only seen in middle and lower panels indicating restoration of endogenous protein expression. Nuclei (DAPI) were pseudo-colored to highlight colocalization puncta. (b) Each bar represents the total percentage of MeCP2 protein foci positive cells relative to DAPI positive cells in each mouse group. Each datapoint represents one microscopic field of view. Non-injected Rett mice show background levels of immunofluorescence44. The distribution of positive nuclei in non-injected wild-type mice reflects the cellular heterogeneity within the brain. Analysis was performed by confocal imaging in three sections from each brain region indicated in the figure. We selected the four brain regions with the highest editing yields. Data is shown as the median percentage of cells showing MeCP2 protein foci ± c.i. (confidence interval 95%) from N = 2 mice per bar, as counted in 3 (non-injected Rett mice) or 6 (injected Rett mice) fields of view per animal. (c) High correlation between the median editing yields (amplicon sequencing) and the percentage of cells with MeCP2 protein foci in the four brain regions in a). The abbreviations stand for Cortex (Cx), midbrain (Mb), thalamus (Th), and brainstem (Bs). Data is shown as one datapoint per brain region. Each datapoint represents the median result of the respective tissue from N = 2 mice injected with targeting virus per readout. For technical reasons the correlated editing yields and protein foci percentages are from different animals.
Extended Data Fig. 10 Proposed mechanisms for the suppressive effect of G·U wobble base pairs on RNA editing when placed at nearest neighbor sites.
(a) Superposition of a 5´ G U wobble base pair (blue) on top of a Watson-Crick AU base pair (black) arranged according to panel b) and c). The shifted Guanine (blue) results in the projection of an exocyclic amino group into the minor groove, which is a distinctive feature of the GU wobble base pair32. (b) hADAR2d/Bdf2 crystal structure indicating the close proximity of the 5’ nearest neighbor AU base pair and Glycine 489. The position of the observed base pair (black, bold) and the target adenosine (green, bold, underlined) is highlighted in the Bdf2 duplex sequence. (c) A 5´ G·U wobble base pair fitted into the hADAR2d/Bdf2 crystal structure at the 5’ nearest neighbor position predicts a steric clash between the exocyclic amino group of the Guanine and the Glycine 489. The clash would require extensive accommodation of the complex and thus cause structural perturbations, that might negatively affect the deamination rate. The position of the observed wobble base pair (blue, bold) and the target adenosine (green, bold, underlined) is highlighted in the Bdf2 duplex sequence. (d) As a) but arranged according to panel e) and f). (e) As b) but showing the 3’ nearest neighbor AU base pairs proximity to Serine 486. The AU base pair was fitted into the position of the original CG base pair of the hADAR2d/Bdf2 crystal structure. (f) As c) but showing the Guanosine of the 3’ G·U wobble base pairs clashing with Serine 486. The 3´ G·U wobble base pair was fitted into the position of the original CG base pair of the hADAR2d/Bdf2 crystal structure.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Reautschnig, P., Fruhner, C., Wahn, N. et al. Precise in vivo RNA base editing with a wobble-enhanced circular CLUSTER guide RNA. Nat Biotechnol (2024). https://doi.org/10.1038/s41587-024-02313-0
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41587-024-02313-0
Discover more from Tamfis Nigeria Lmited
Subscribe to get the latest posts sent to your email.











