
 Hot Deals
Hot Deals Shopfinish
Shopfinish Shop
Shop Appliances
Appliances Babies & Kids
Babies & Kids Best Selling
Best Selling Books
Books Consumer Electronics
Consumer Electronics Furniture
Furniture Home & Kitchen
Home & Kitchen Jewelry
Jewelry Luxury & Beauty
Luxury & Beauty Shoes
Shoes Training & Certifications
Training & Certifications Wears & Clothings
Wears & Clothings





28
May
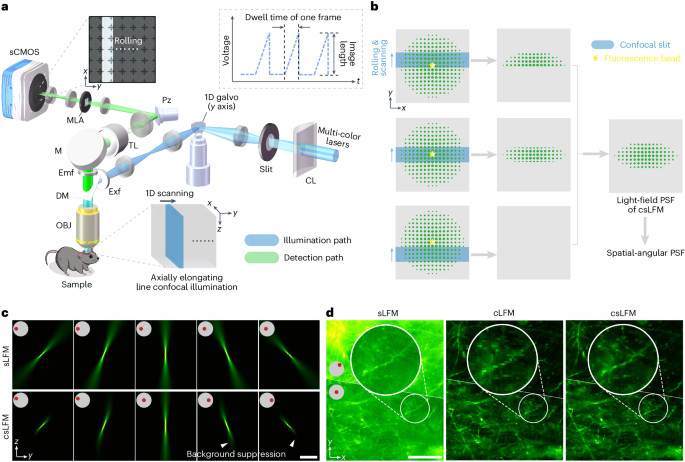
Long-term intravital subcellular imaging with confocal scanning light-field microscopy
Technology tamfitronics MainIntravital imaging1,2,3,4,5,6,7 is vital for studying diverse physiopathological proce...
You must be 18 years of age or older to view page. Please verify your age to enter.
Your access is restricted because of your age.
Please confirm you want to block this member.
You will no longer be able to:
Please note: This action will also remove this member from your connections and send a report to the site admin. Please allow a few minutes for this process to complete.