
 Hot Deals
Hot Deals Shopfinish
Shopfinish Shop
Shop Appliances
Appliances Babies & Kids
Babies & Kids Best Selling
Best Selling Books
Books Consumer Electronics
Consumer Electronics Furniture
Furniture Home & Kitchen
Home & Kitchen Jewelry
Jewelry Luxury & Beauty
Luxury & Beauty Shoes
Shoes Training & Certifications
Training & Certifications Wears & Clothings
Wears & Clothings





05
Nov
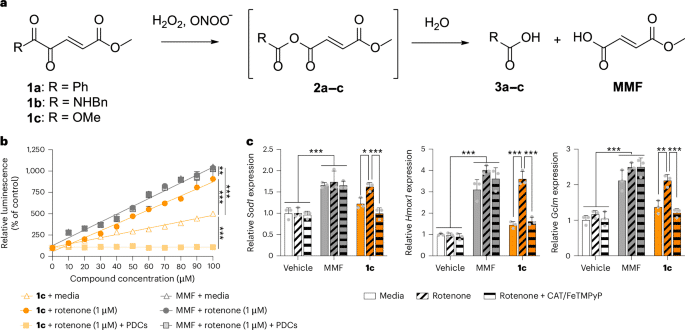
Site-specific drug release of monomethyl fumarate to treat oxidative stress disorders
-
Posted by
 Tamunofiniarisa
Tamunofiniarisa
Technology tamfitronics MainThe transcription factor nuclear factor E2-related factor 2 (NRF2) is an attractive the...